November 4th, 2025
Enabling new AWS families with GPU-enabled Instances on Data Studio
This release introduces adding new AWS families with GPU enabled Instances within Seven Bridges Data Studio. It enables users to perform advanced medical image visualization, segmentation, GPU acceleration and secure data integration.
Enabled AWS Instances on Data Studio
We are thrilled to announce that we have enabled new AWS families with GPU enabled Instances on Data Studio to provide even more options for optimizing your computational workloads.
New AWS Instance Families Include:
- Compute-Optimized: C7i, C6i
- Memory-Optimized: R7i, R7iz, R6i, R6in, X2idn, X2iedn, X2iezn
- General Purpose: M7i, M6i
- GPU-Enabled: G6, G5
This update empowers researchers and analysts to execute high-performance computational tasks directly from Data Studio without external infrastructure setup.
It simplifies workflow execution, reducing processing time and improving efficiency for GPU-dependent applications.
November 4th, 2025
3D Slicer now available in Data Studio
General
This release introduces 3D Slicer as a fully integrated analysis environment within Data Studio, enabling advanced medical image visualization and segmentation.
New feature
3D Slicer can now be launched directly from Data Studio for interactive medical imaging and 3D visualization workflows, with the option of GPU-powered compute instances for high-performance rendering. This release enables you to perform entire imaging workflows from data import, visualization, annotation within the secure and compliant Seven Bridges platform, and eliminates the need to move sensitive data across multiple tools or local machines.
October 30th, 2024
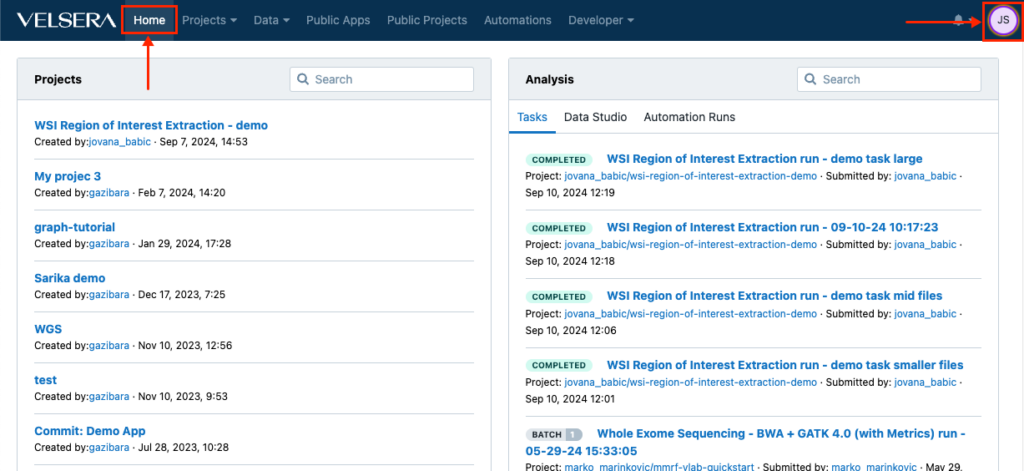
Changes in the main navigation
The main navigation now features a Home button which returns you to the dashboard at any given point. In addition, the icon for accessing the account settings now shows your initials instead of the username.
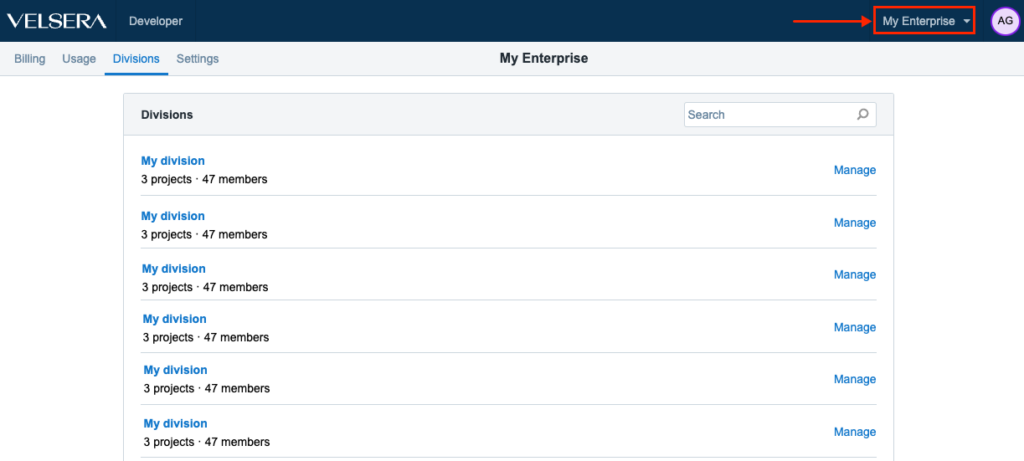
Improved UX for Enterprise users
We have introduced a small change in order to improve and optimize the UX for the Enterprise users. The menu for accessing Divisions is now in the upper right hand side, right next to the Account Settings.
October 28th, 2024
Nextflow – enhanced execution and scalability
We’re excited to announce powerful new features to enhance Nextflow performance, scalability, and resource optimization. These updates streamline workflow execution and expand support for advanced configurations, making it easier than ever to manage complex pipelines.
Multi-Instance Execution
Support for multi-instance execution allows Nextflow workflows to leverage multiple instances concurrently. This improvement enables increased parallelism and better resource utilization across workflows, resulting in significant performance boosts for complex or large-scale data pipelines.
Memoization (Work Reuse)
Introducing memoization for workflow tasks, allowing Nextflow to reuse previous results, significantly reducing compute time and resource usage. This feature is especially beneficial for iterating on workflows with minor changes, as previously executed tasks are reused when input data and parameters remain consistent.
Elastic Disk Support
Elastic disk capability allows dynamic scaling of disk resources based on workflow requirements. This flexible storage management ensures that high-demand workflows have access to the necessary storage without manual intervention, optimizing both performance and cost.
Instance Hints for Optimized Resource Selection
Nextflow now supports instance hints to provide tailored recommendations for instance types based on workflow needs. These hints help reduce costs and improve runtime efficiency by selecting the most suitable instance types for different tasks.
Real-Time Job Monitoring for Worker Instances
Enhanced job monitoring capabilities provide real-time insights into the status of worker instances. Users can monitor task progress and troubleshoot issues as they arise, leading to faster adjustments and a more seamless workflow execution experience.
Compatibility with All Nextflow Executor Versions (Post 21.10.0)
Full support for all Nextflow executor versions after 21.10.0 allows users to run workflows with the exact executor version they need, ensuring compatibility and reproducibility across executions. This enhancement allows teams to replicate their work consistently while taking advantage of continuous improvements in Nextflow ecosystem.
June 10th, 2024
Recently published apps
Somatic small variant callers for long read data, ClairS (0.2.0) and ClairS-TO (0.1.0) (for matched tumor-normal pairs and tumor-only data, respectively) have been published to the Seven Bridges Platform.
May 13th, 2024
Recently published apps
We have published the GCTA 1.94.1 tool on the Seven Bridges Platform. GCTA is a suite of tools for various genetic analyses using genome-wide data. GCTA (Genome-wide Complex Trait Analysis) was initially developed to estimate the proportion of phenotypic variance explained by all genome-wide SNPs for a complex trait but has been greatly extended for many other analyses of data from genome-wide association studies (GWASs)
May 10th, 2024
Recently published apps
snM3C pipeline
The snM3C pipeline is designed for profiling 3D genome structure and DNA methylation in single cell data as a part of the Human Cell Atlas and the WARP BRAIN Initiative.
The snM3C pipeline performs:
- Demultiplexing (by the Demultiplexing custom tool)
- Reads sorting (by the Sort custom tool)
- Reads trimming (by Cutadapt)
- Paired-end reads alignment (by Hisat-3n)
- Separating unmapped, uniquely aligned, and multi-aligned reads (by Separate unmapped reads wrapped around a custom script)
- Splitting unmapped reads by enzyme cut site (by Split unmapped reads wrapped around a custom script)
- Alignment of the unmapped, single-end reads (by Hisat-3n)
- Removing the overlapping reads (by Remove overlap read parts wrapped around a custom script)
- Merging mapped reads from single- and paired-end alignments (by Samtools Merge)
- Removing duplicate reads (by Picard MarkDuplicates)
- Calling chromatin contacts (by Call chromatin contacts wrapped around the custom script)
- Creating ALLC files (by Allcools bam-to-allc)
- Creating summary output (by Allcools extract-allc)
All tools are wrapped for the workflow specifically and use retagged us.gcr.io/broad-gotc-prod/m3c-yap-hisat:1.0.0-2.2.1 Docker image.
DeepSomatic 1.6.1
DeepSomatic is an extension of DeepVariant for calling somatic variants from matched tumor-normal data. The tool is still in active development and only WGS data is currently supported.
SortMeRNA 4.3.6
SortMeRNA is a local sequence alignment tool for filtering, mapping and OTU clustering. The main applications of SortMeRNA are filtering rRNA from metatranscriptomic data, OTU-picking and taxonomy assignation available through QIIME v1.9+.
dupRadar 1.32.0
The dupRadar tool is intended for duplication rate quality control for RNA-Seq data. It gives an insight into the duplication problem by graphically relating the gene expression level and the duplication rate present on it.
April 8th, 2024
Recently published apps
Here are the new apps published in our Public Apps gallery:
- ASCAT 3.1.2 tools (ASCAT prepareTargetedSeq, ASCAT prepareHTS and ASCAT). ASCAT prepareTargetedSeq prepares SNP references for ASCAT processing of targeted sequencing data. ASCAT prepareHTS prepares sequencing data (WGS, WES or targeted) for ASCAT. ASCAT infers tumor ploidy, purity and allele-specific copy number profiles.
- JAFFAL 2.3 tool. JAFFAL is used to detect fusion genes from long-read (PacBio and ONT) transcriptome sequencing with high accuracy, overcoming the challenges posed by higher error rates in long-read data.
- Ballgown 2.34.0 toolkit. Ballgown is a package designed to facilitate flexible differential expression analysis of RNA-Seq data. It also provides functions to organize, visualize, and analyze the expression measurements for transcriptome assembly
Apps with version updates
- StringTie 2.2.1 toolkit. StringTie is a fast and highly efficient assembler of RNA-Seq alignments into potential transcripts. StringTie Merge tool merges/assembles GTF/GFF transcript files into a non-redundant set of transcripts. This tool should be used after StringTie transcript assembling of each sample in the experiment.
April 1st, 2024
Recently published apps
We published the following ASCAT 3.1.2 tools in our Public Apps gallery:
- ASCAT prepareTargetedSeq prepares SNP references for ASCAT processing of targeted sequencing data.
- ASCAT prepareHTS prepares sequencing data (WGS, WES or targeted) for ASCAT. ASCAT infers tumor ploidy, purity and allele-specific copy number profiles.
Recently updated apps
We updated the following apps from the MSIsensor v0.6 toolkit:
-
MSIsensor scan – a tool for cataloging homopolymers and miscrosatelites sites in the reference genome. It prepares reference for MSIsensor msi.
-
MSIsensor msi – a tool for somatic microsatellite changes detecting and scoring. Designed to work with paired tumor-normal data.