Optimizing novoBreak on the Cancer Genomics Cloud
Last week, we released novoBreak—an exciting new bioinformatics software—on the Cancer Genomics Cloud (CGC). The tool, described by Zechen Chong and colleagues in Nature Methods, is a novel approach to detect breakpoints in cancer genomes with high precision and sensitivity.
In this post, we explain how Zechen used the Publish your app feature of the CGC to make novoBreak widely accessible to the research community, and how our bioinformaticians worked with Zechen to optimize novoBreak’s cost and runtime.
Chromosomal breakpoints to detect and target cancer
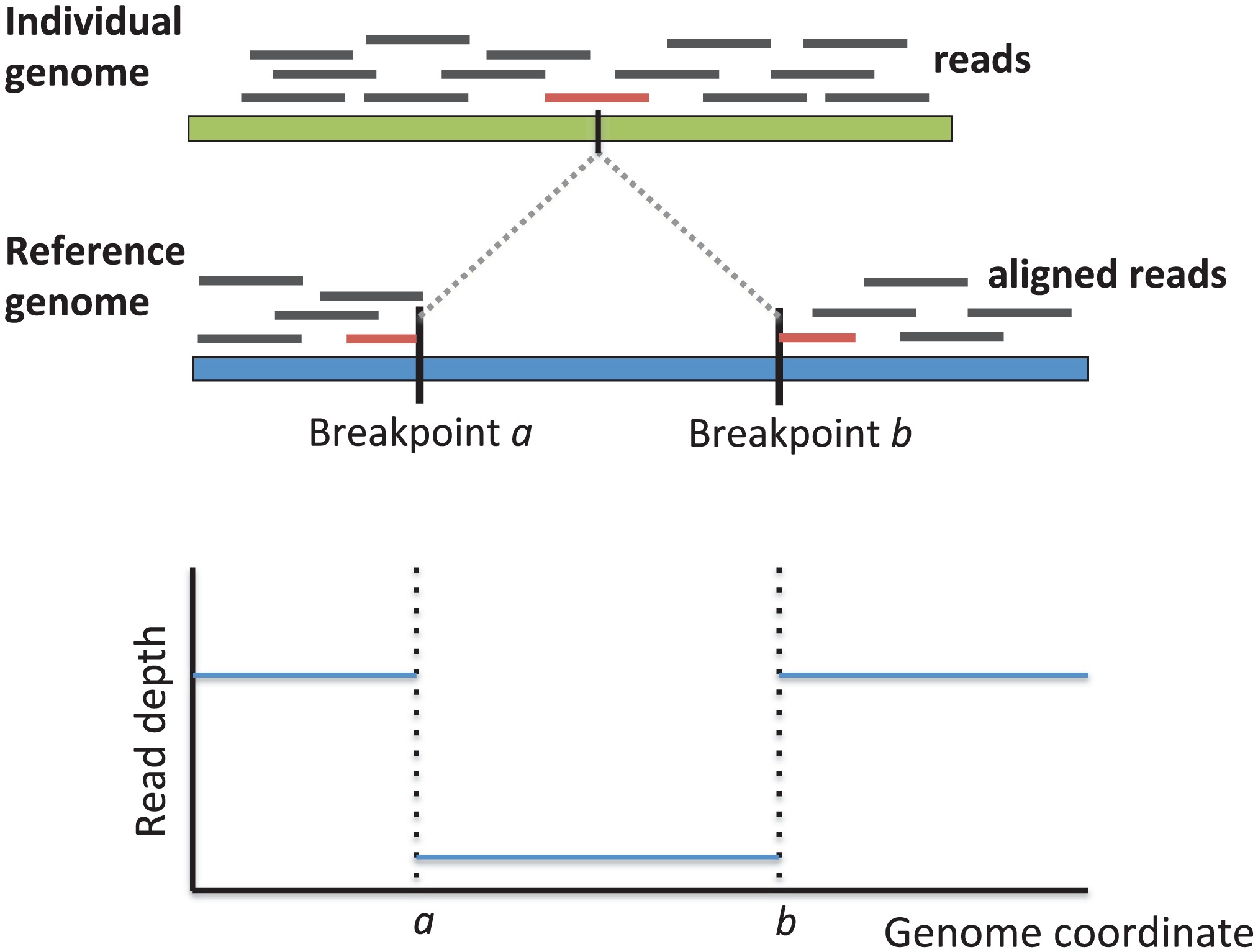
Breakpoints are locations on the chromosome where translocations occur during recombination. Translocations can play a role in disease when they result in gene alterations or dysregulation. In particular, a range of translocations and their resulting gene fusions have been linked to different cancer subtypes.
With the proliferation of next-generation sequencing technologies, researchers are able to examine structural variation in cancer genomes with increased throughput. Numerous approaches for detecting structural variants use DNA short read data to identify regions with discordant read pairs, split reads, or read depth anomalies. However, these approaches are limited by their reliance on read alignment accuracy, which is poor when reads deviate from the reference genome.
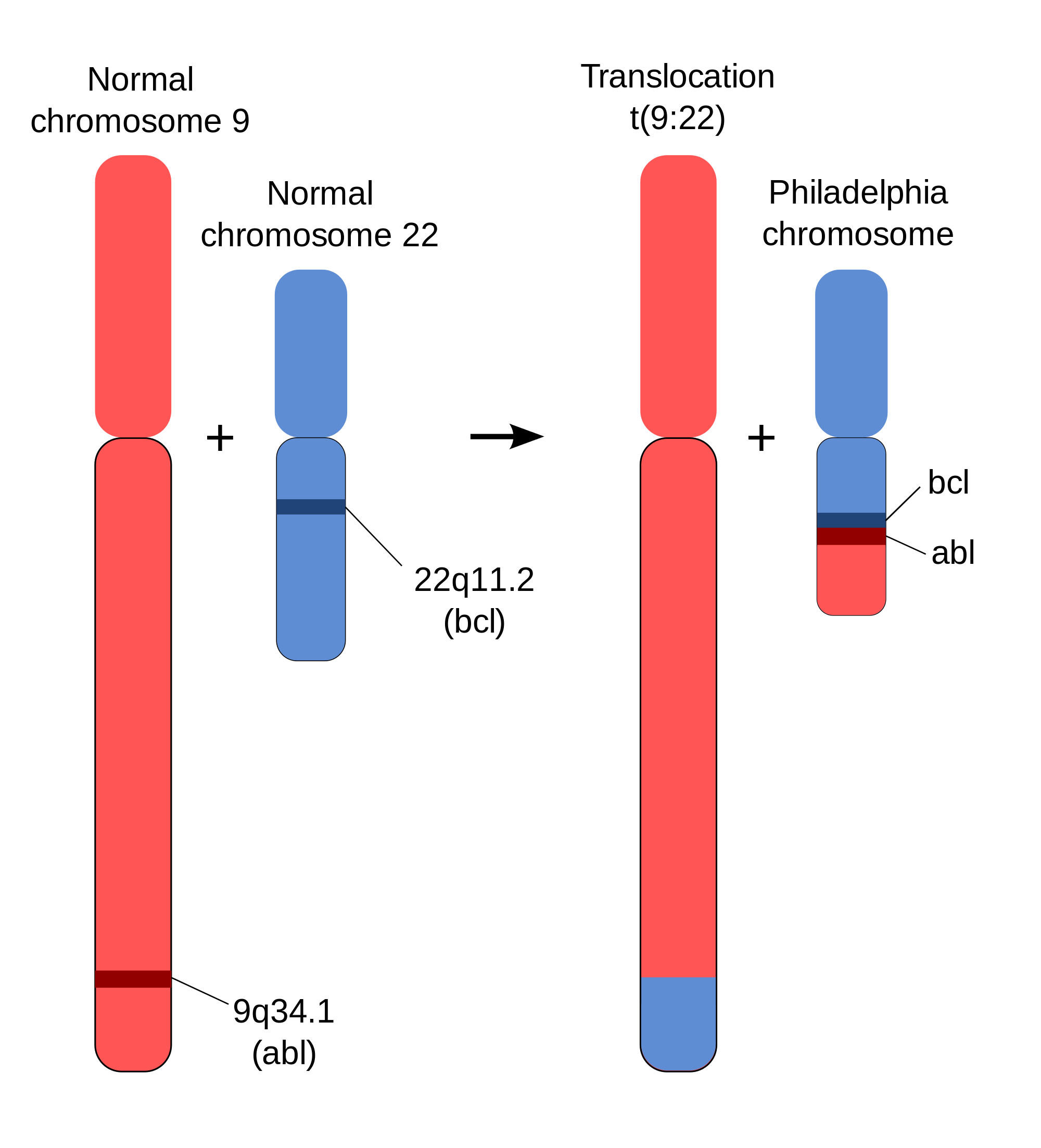
The prototypical example of a structural variant linked to cancer is the contribution of the Philadelphia chromosome to chronic myeloid leukemia. A translocation between chromosomes 9 and 22 results in an altered chromosome 22, called the Philadelphia chromosome, along with the gene fusion BCR–ABL. The gene fusion produces an oncogenic, constitutively active tyrosine kinase. As the gene fusion is present in over 90% of patients with chronic myeloid leukemia, a genetic test to identify the gene fusion is used for clinical diagnosis of the disease. Chemotherapeutics such as imatinib have been developed to target the fusion protein.
novoBreak for breakpoint detection in cancer genomes
novoBreak is designed to enhance detection of structural variant breakpoints. To improve alignment of tumor DNA to the reference genome, the novoBreak algorithm divides tumor reads into shorter sections that are k nucleotides long (k-mers). By filtering out k-mers that match the reference and normal genomes, the algorithm identifies k-mers that are unique to the tumor and indicate breakpoints. Reads containing the unique k-mers are assembled into contigs local to the breakpoints, which are then aligned to the reference genome in order to infer exact breakpoints and their associated structural variants.
When novoBreak was tested on synthetic tumor data from the ICGC-TCGA DREAM 8.5 Somatic Mutation Calling Challenge, it matched top-performing tools (including DELLY and Manta) in terms of precision and outperformed them in sensitivity. When tested on whole genome sequencing data from a melanoma cell line, novoBreak identified the most structural variant breakpoints that had been previously validated with PCR and Sanger sequencing.
With novoBreak’s improvement in structural variant detection, researchers can use the tool to identify novel gene fusions relevant to cancer.
Publishing novoBreak on the CGC
Zechen used the Publish your app feature of the CGC to make novoBreak broadly available and useful to the research community. He first installed the software in a Docker container, and then used the CGC’s visual tool editor to describe novoBreak in the Common Workflow Language, the specification for describing analysis tools and workflows that makes them portable and scalable across environments.
With novoBreak hosted on the CGC, researchers can immediately access and run it without downloading or configuring software. Importantly, the CGC hosts key public cancer genomics data sets, including The Cancer Genome Atlas, Cancer Cell Line Encyclopedia, and Simons Genome Diversity Project, which researchers can immediately analyze with published tools such as novoBreak.
Optimizing novoBreak
When Zechen initially installed and described novoBreak on the CGC, running the software had a relatively high computational cost, limiting its widespread use by researchers. At his request, Seven Bridges bioinformaticians worked with Zechen to understand the novoBreak workflow and identify how it could be optimized.
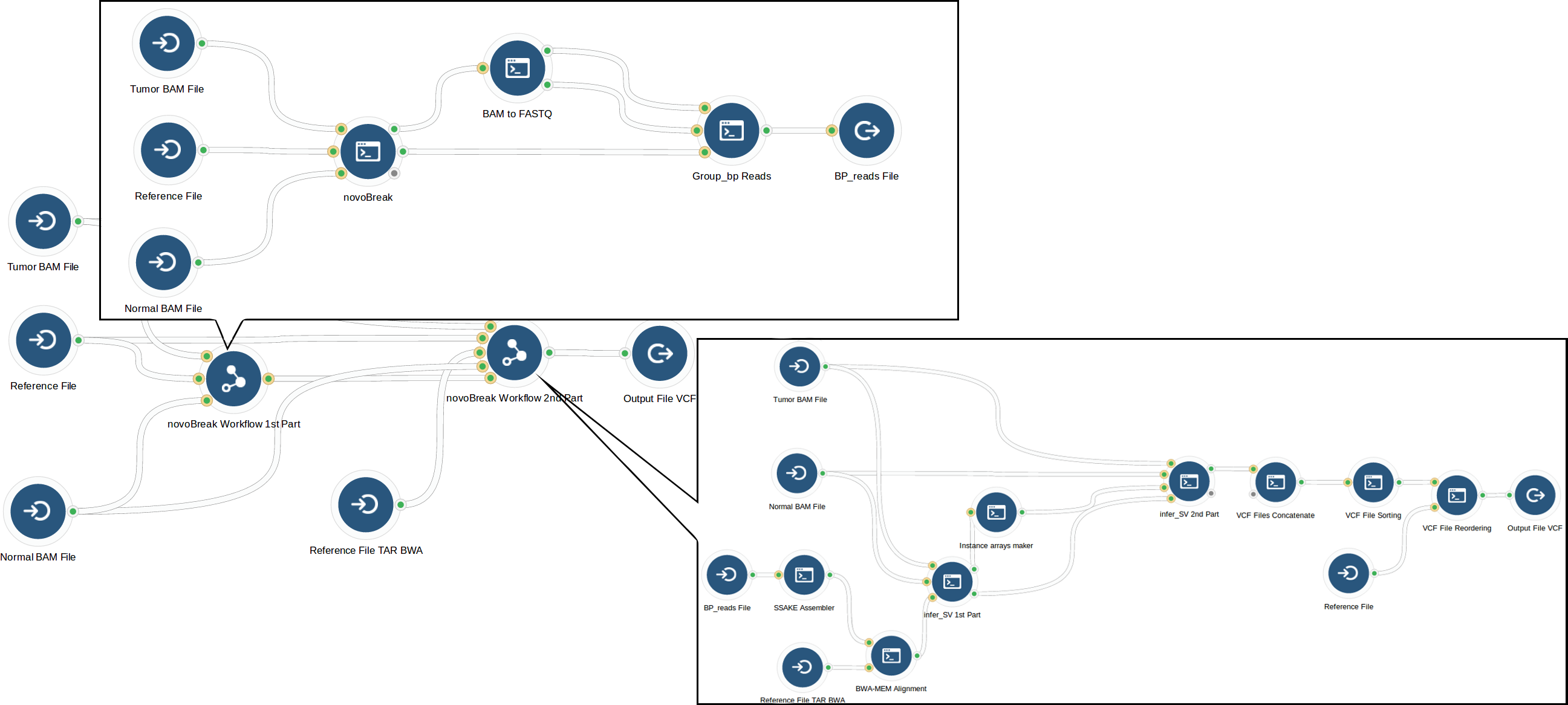
After analyzing the novoBreak workflow, our bioinformaticians optimized it using whole genome samples from The Cancer Genome Atlas. The initial part of the workflow that created k-mers from reads was memory intensive, so they determined the optimal computation instance that met novoBreak’s CPU and memory requirements while minimizing cost. This optimization decreased the run cost; however it also increased runtime substantially (to more than 4 days). In response, our bioinformaticians focused on the part of the novoBreak workflow that identifies breakpoints, where large numbers of files were processed sequentially. When they modified the step to process files in parallel, runtime was drastically reduced. Through these optimizations, they reduced run cost by 40% while keeping runtime to around 1 day.
In the end, our team helped Zechen to make novoBreak not just widely available, but also immediately usable by the research community to advance our understanding of structural variants in cancer.
“CGC has tremendous capability for executing our tool, novoBreak, on a large cohort of tumor samples for structural variations analysis, which is an impossible mission on institutional level high performance computing (HPC) clusters. We are very happy to see novoBreak deployed as a public workflow by CGC team and we expect novoBreak can contribute more to the community through the CGC platform.”—Zechen Chong
Researchers can access the optimized novoBreak tool on the CGC, in the Public Apps gallery. See the CGC Knowledge Center for detail of how to publish your own software.
The optimization project was led by Raunaq Malhotra, Bioinformatics Scientist at Seven Bridges. Email team@sevenbridges.com to talk about how to publish and optimize your tools on the CGC.