The Cancer Genome Atlas
The Cancer Genome Atlas (TCGA) is the world’s largest and richest collection of genomic data. It is a comprehensive and coordinated effort to accelerate our understanding of the molecular basis of cancer.
There are around 200 types of cancer, each characterized by molecular changes of the genome. These changes can be identified if:
- Large amounts of genomic data are available to analyze;
- All the data is easily accessible;
- Researchers collaborate to develop bioinformatic and mathematical tools; and
- Enough computing power is available to analyze the data.
Data collection for TCGA began as a joint effort by the National Cancer Institute (NCI), National Human Genome Research Institute (NHGRI), National Institutes of Health (NIH), and the Department of Health and Human Services.
TCGA currently contains more than 2.5 petabytes of publicly available data, which have contributed to hundreds of cutting-edge cancer studies.
What’s inside?
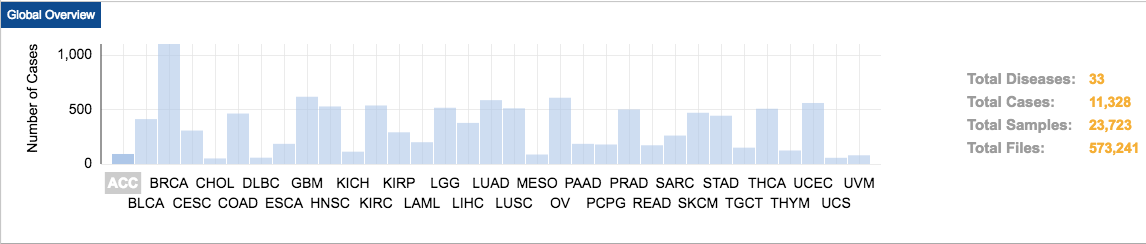
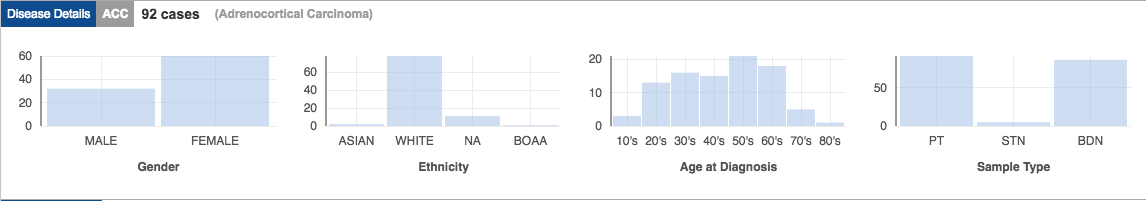
TCGA contains data on 33 different cancer types from 11,328 patients. These cancer types were chosen because of their poor prognosis and availability of samples. Matched tumor–normal tissue samples are molecularly characterized to identify genomic alterations.
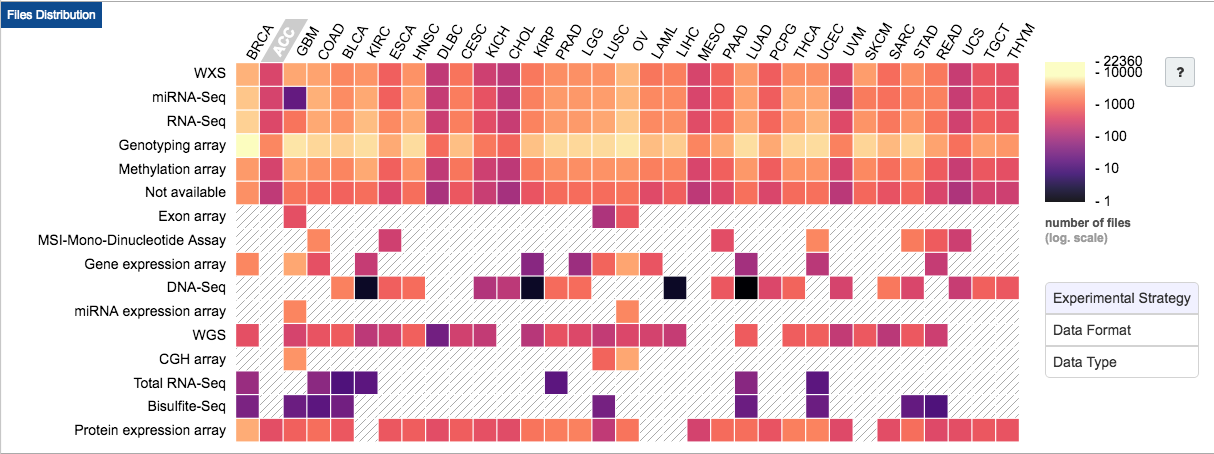
TCGA contains molecular data from multiple types of analysis:
- DNA sequencing
- Whole genome sequences
- Whole exome sequences
- Sequence traces
- Mutations, including coding, splice site, germline and noncoding somatic variants
- RNA sequencing
- miRNA sequences (calculated expression per miRNA and isoform)
- mRNA sequences (calculated expression per gene, exon, splice junction, isoform)
- Total RNA sequences (calculated expression per gene, exon, splice junction, isoform)
- Expression signals per gene, exon, splice junction, miRNA, and isoform
- Copy number
- Arrays (raw, unnormalized, normalized)
- Low-pass DNA sequencing (whole genomes sequences, variants, coverage)
- Array-based expression
- Gene expression (raw, normalized, calls)
- Exon expression (raw, normalized, calls)
- miRNA expression (raw, normalized, calls)
- DNA methylation
- Bisulfite sequencing (whole genome sequence, methylation and mutation calls)
- Array-based methylation (raw signal intensity, calculated beta values)
- Other
- Protein expression (high-resolution images of protein arrays, raw signals, normalized expression)
- Microsatellite instability (markers, classifications)
In addition to molecular data, TCGA has well cataloged metadata for each sample:
- Clinical information about participants (e.g., sex, race, ethnicity, drugs taken, and response to treatment);
- Information about the samples (e.g., the weight of a sample portion, days to collect, and time of freezing); and
- Images of the tumors (allows estimation of number of proliferating cells, how many cells have died, how many immune cells are present, etc.).
Accessing the data
TCGA data is available in two tiers:
- Open Access: Public data not unique to an individual and not requiring user certification. These data include deidentified clinical and demographic data, gene expression, copy number alterations, epigenetic data, compiled summaries, and anonymized single amplicon DNA sequence data.
- Controlled Access: Data that can be unique to an individual (stripped of direct identifiers), requiring user certification. These data include primary sequence data, SNP array data, exon array data, VCFs, and certain information from MAFs.
TCGA data is available to download, but it would take several weeks to download all the data with standard internet speeds. Once downloaded, the data then costs a substantial amount to store and requires powerful computational resources to explore, manipulate, and analyze.
An alternative data access model takes advantage of the capabilities of the cloud to support researchers working with TCGA data. The Seven Bridges Cancer Genomics Cloud (CGC; an NCI Cancer Genomics Cloud pilot) hosts petabytes of TCGA data alongside hundreds of bioinformatic tools, and gives on-demand access to thousands of CPU cores for analysis.
Researchers can register at www.cancergenomicscloud.org to instantly explore and work with TCGA, the world’s most-complete public cancer genomic dataset.