Assessing State of the Art Bioinformatics
The Oxford English Dictionary defines bioinformatics as “the science of information and information flow in biological systems, especially of the use of computational methods in genetics and genomics.” In common vernacular, it is often defined as the use of statistical and computing methods to solve or better understand complex biological problems. In 1990, the Human Genome Project started with a goal of sequencing the entire genome of one human individual for the first time in history. As the project progressed it became evident that substantial computational power and algorithms would be needed to enable faster and more accurate reconstruction of the genomic sequence. After a total investment of 13 years and 3 billion dollars, the successful end of the project in 2003 marked the birth of a new discipline: bioinformatics. In the years to follow, the exponential fall of sequencing costs enabled by development of Next Generation Sequencing (NGS) technology contributed to a similarly exponential growth of the number of sequenced samples. Necessity is the mother of invention, and in the past several years there has been a vast array of tools and algorithms developed by the research community to meet the need for fast and accurate sequence analysis. A majority of different analytical processes have dedicated tools, and others even have dozens of alternative tools that could be used.
As time passed by, with projects and experience multiplying, and collaborations with many researchers involved in studies that include larger cohorts, the major challenges emerged. It appeared that most of the time spent on projects was not in implementing or understanding algorithms. In fact, in most projects the time spent on algorithms was neglectable. Major time was spent on constructing the analysis using already implemented algorithms, and adjusting (optimizing and validating) them to be able to process a large number of samples in a reasonable time and at a reasonable price. So, the question emerged: what is the state-of-the-art bioinformatics?
Bioinformatics analysis usually considers executing a number of bioinformatics tools that pass the results from the first to the second tool, from second to third, and so on. Many times the number of tools is large and their execution flow is not sequential but rather graph-like. To be more precise, the flow forms a directed connected acyclic graph commonly known as a bioinformatics workflow (pipeline). An example of such a workflow is shown in Figure 1.
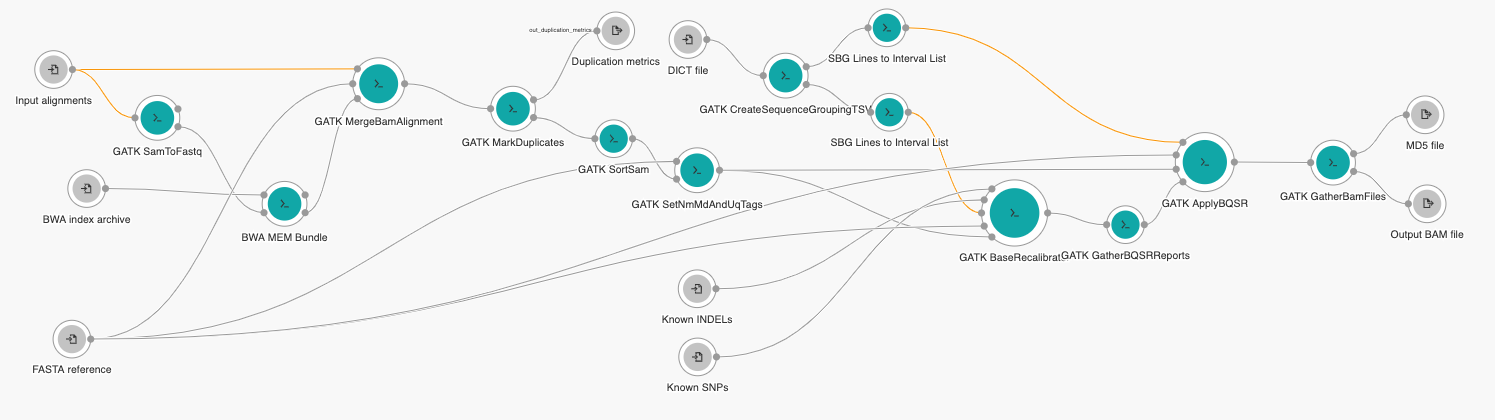
Two current standards that enable portable and reproducible execution of bioinformatics workflows (hereafter just “workflows”) are: Common Workflow Language (CWL) and Workflow Description Language (WDL). No need to waste words on why it is extremely important to define the workflow very precisely and to be able to obtain the same results after running the workflow on the same or different environment: reproducibility and portability. It is extremely important to define the workflow precisely in order to ensure reproducibility and portability when running the workflow in different environments. Besides the exact description, another feature that greatly contributes to reproducibility and portability is the fact that the tools can be executed in the predefined virtual environment: Docker images. In that way the produced result will not depend on the platform or host operating system where the tool is executed.
Another widely-present demand generated by the huge amounts of sequenced samples is execution on the almost unlimited cloud resources. Since more and more researchers (often without deep technical knowledge) are getting involved in genomics, there is a need for creating an easy-to-use and flexible environment (web platform) for storing genomics data and processing them on the cloud. All necessary information for executing the workflow on the cloud is defined inside the workflow description. That includes the location of the input files, docker images, bioinformatics tools that should be executed, necessary computational resources and location to store the results (Figure 1).
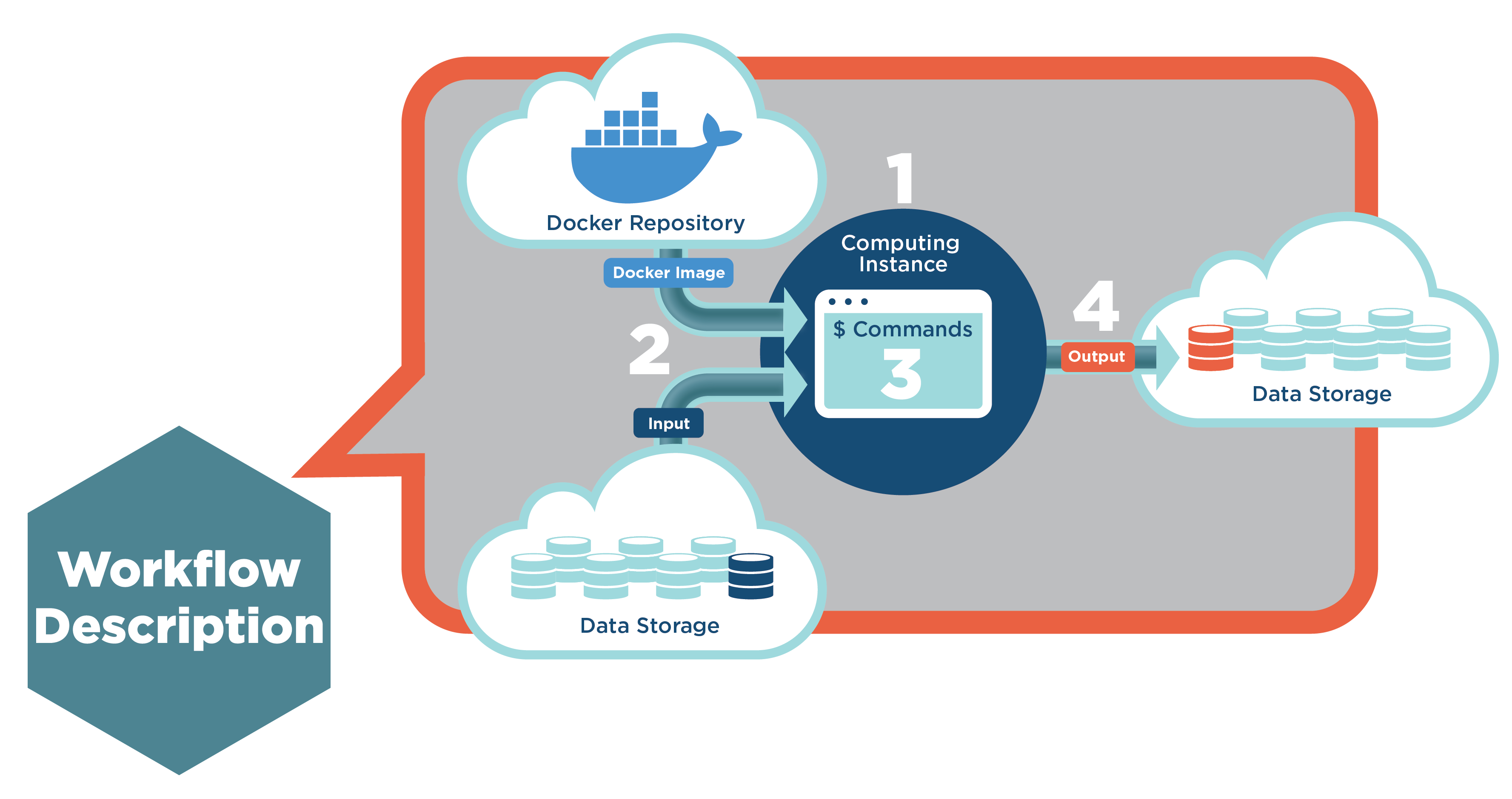
With that defined infrastructure, the only thing left to be done is constructing and optimizing the workflows. While this seems deceptively simple, it can become exceedingly complicated when large numbers of tools with many parameters are utilized in a given workflow. We often use the analogy for constructing the workflow and setting parameters: doing the task with 749 pitfalls (every time a different “random” number is used because it is never known in reality what is the value) and if you fall into any of these, the task will fail. If it fails, this presents a better scenario than if the task completes with the hidden error.
Constructing workflows is a piece of cake, right?
After describing each of the bioinformatics tools to be used in workflow description language, one can start with constructing a workflow. So, what else could complicate the construction of workflows? The selection of tools, adaptation of file types between tools, assigning adequate computational resources, input files mapped to the different reference genomes, and the specific feature of the tool not working as documented are all factors which can complicate workflows. Herein, we will examine each in depth and work through some examples.
Selection of tools
Usually, the first step for the creation of the workflow is the selection of command-line bioinformatics tools that will be included. Typically, the same operation (part of the analysis) that is commonly used in a given workflow can be done with many publicly available tools. One must do the research to decide which one fits the project demands the best. The demands vary but usually include some of the following: high accuracy, small execution time (low cost of the execution), and/or that the tool to be used must be recommended by some institution or validated in some other previous project. An example of the last-mentioned is the Broad Best Practice for DNA Short variant discovery or RNA Seq short variant discovery.
A good example of a task for selection of the tool is deciding which structural variant caller to use. Structural variants are changes in the genome with more than 50 nucleotides in size. They can be very complex and include deletions, insertions, translocations, inversions, and multiplications to the genome, and combinations thereof. This complexity can make variant calling difficult, especially with short Illumina reads. Fortunately, many bioinformatics tools are developed to tackle this challenge. Last year, the Genome In a Bottle Consortium published a truth set structural variants for several sequenced samples, enabling the creation of reliable structural variant callers benchmark (Figure 2). With quantitative confirmation, it is easy to decide which variant caller performs more accurately for different types and sizes of structural variants. Also, when considering only one type of variant (e. g. deletions) we should not pick up a variant caller which is incompatible with other types (e. g. translocations) because these two variant types could coexist at the same location in some cases (e.g. translocation with deletion).
Another example for the selection of the tool is removing duplicates from sequenced DNA reads. It is a common situation that during the process of sequencing a single DNA fragment is duplicated several times with polymerase chain reaction (PCR). During the process of the genome reconstruction, it is good practice to exclude all but one identical copy of this fragment. Several tools are available for this purpose, but choice mostly comes to GATK4 (previously Picard) RemoveDuplicates which removes also optical duplicates. This tool is also recommended by Broad’s Best Practices for DNA short variant discovery. Its only caveat is low speed, as it is single-threaded. There are multi-threaded solutions that are much faster (Biobambam2, Sambamba MarkDuplicates) but they do not remove optical duplicates and are not recommended by Broad’s Best Practices. So, the decision is to be made in terms of desired priority: big gains in execution time versus eventual small accuracy changes and avoiding standards.
Adaptation of file types between tools
When the main players are selected, the next step is to choose the tools that bridge their interfaces and files that they exchange. Many examples of this adaptation exist, probably the most present is the conversion of SAM alignment format to BAM. Most of the aligners output file in SAM format (BWA-MEM, Bowtie2), but the tools that follow it in the analysis require its binary representation: BAM format. This includes the tools for correction of the base qualities (GATK4 BaseRecalibrator), extracting alignment metrics (GATK4 CollectAlignmentSummaryMetrics), etc. These tools also require an index file (BAI) available together with BAM. To meet these two requirements the workflows should be equipped with adaptation tools: SAM to BAM converter and BAM indexer tool.
Assigning adequate computational resources
Some of the workflows can be hungry for certain computational resources, and since usually the resources become limited, workflows cannot be executed. Computational resources include the number of processors available (with only one CPU used some workflows can run for weeks), available memory, (with lack of memory some tools cannot complete the execution) and hard drive space allotted (with a deficit of space storing of the input or output files is not possible). Assigning minimal required resources for workflow execution often represents a real challenge.
Dependency between execution time and the number of processors is usually not linear and it is closer to logarithmic, eventually reaching saturation (assigning additional processors to the tool does not lower its execution time). It is also a common situation that several (multi-processor) tools could be executed simultaneously. Therefore, ideally the number of processors to use at each moment should be assigned to every tool in order to obtain maximal usage of the available resources and minimal execution time.
Typically, the amount of required memory for successful tool execution increases with the number of processors used and input file size. Unfortunately, memory recommendations for the majority of the tools are not given, and need to be tested manually. When tests are conducted, it is useful to make them publicly available for other users, so the majority of the tools in the Seven Bridges Public App Gallery contain benchmarks in the description with the required memory footprint.
Determining sufficient hard drive space to assign can be done by summing the sizes of all input, intermediate (files produced inside the workflow and passed to the other tools but not on the output of the workflow) and output files of the workflow. Since the workflows could be very complex it can be difficult and error-prone to predict minimum sufficient hard drive space, even with neglecting smaller files.
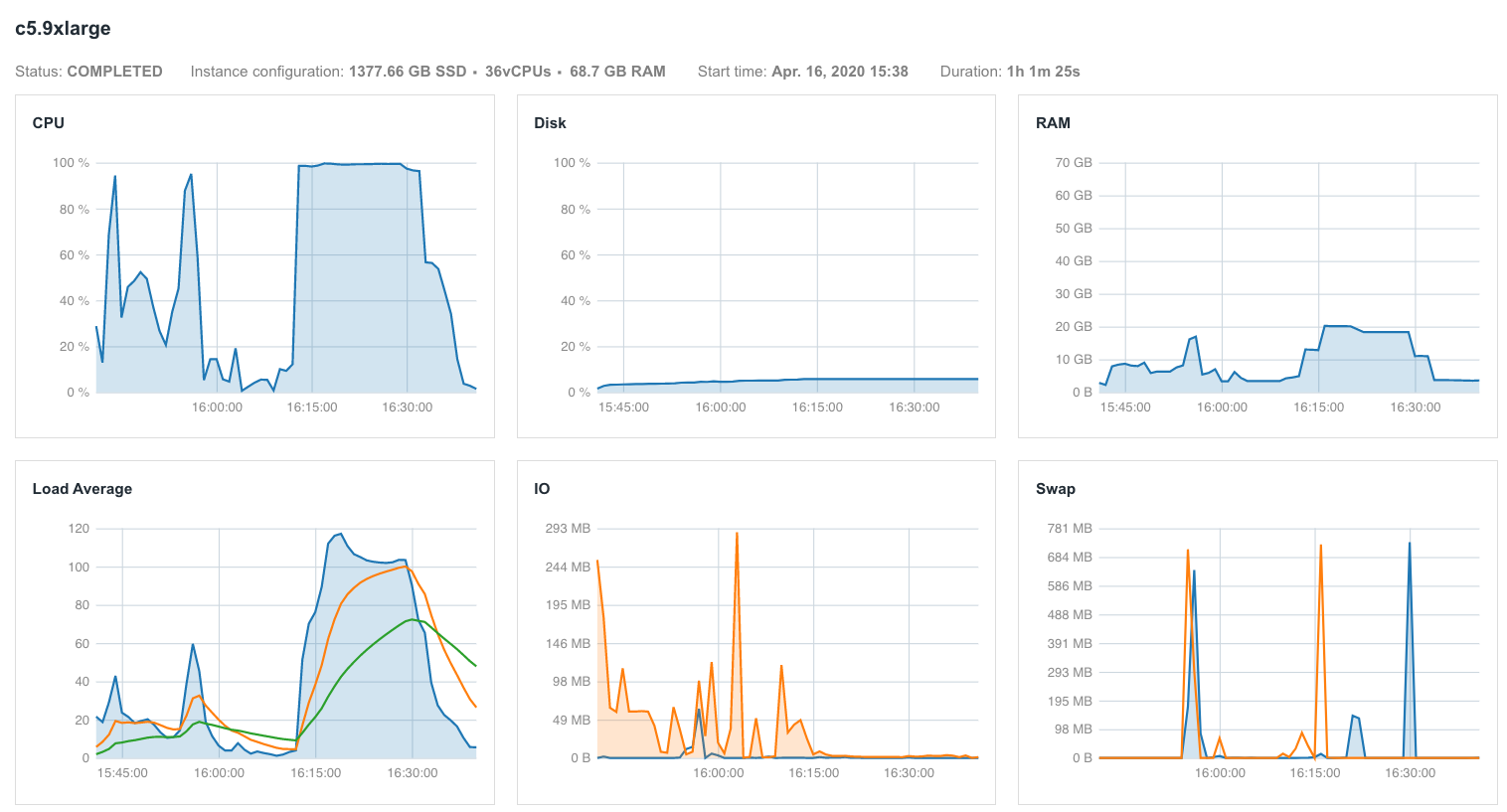
In an ideal world, the automatic scheduling of all resources could be performed. Up until now, there are no publicly available automatic schedulers for bioinformatic workflows. The setting of these three resources is usually done by initially assigning larger values than required and then running a test for several different input types or sizes while measuring the usage during the execution (Figure 3). After successful completion, the adjusting of resource allotment is done by selecting the machine that satisfies these minimum requirements.
Chromosome names in reference genome: chr1 vs. 1
The most-used builds of the human reference genome originate from two institutions: Genome Reference Consortium (GRC) and University of California Santa Cruz (UCSC). Chromosomal sequences between them are the same, and if we put aside smaller differences in mitochondrial regions what is left is different chromosome names. GRC uses chromosome names “1”, “2”, “3”, etc, while UCSC “chr1”, “chr2”, “chr3”, etc. All reference databases and other files used as inputs to the workflow are aligned (mapped) to the reference genome, and it is necessary for all the inputs to the workflow to be mapped to the same one. It is a very common situation that, for example, reference databases are mapped to the GRCh38 reference genome and the workflow requires the usage of the UCSC hg38. This problem sounds trivial, and it actually is. However, this situation and ones like it occur so often that the human and computational time spent on these conversions to date could be measured in the hundreds of millions of dollars. Luckily, a good explanation on which reference genome to use is available.
Specific feature of the tool not working as documented
Most of the publicly available bioinformatics tools are developed by scientists, usually during their Ph.D. studies, and few are developed by institutions with software development teams. Frequently, tools so developed are not thoroughly tested, so using it with configuration not considered by the author may cause undesired behavior or trigger a bug. When this happens we should be happy for the received opportunity to “give back to the community” by submitting a Github issue (most of the tools are distributed through Github repositories) in the repository of the tool. Of course, most of us are not happy with facing unreported tool’s errors. Nevertheless, we should be extra polite and keep in mind the big favor the author of the tool did for us: providing the coding and support for the tool, free of charge. Even a bigger gift for the community we can give by fixing the tool ourselves and submitting the pull request on Github. Sometimes, when waiting for the change that is too complex for us to implement is not acceptable, we could change the configuration of other tools to attempt to avoid potential degradation of output quality. To completely avoid these situations, we should be more considerate during the process of selecting the tool, at least by looking at the number of issues present for it (it correlates directly with the number of users of the tool), and response time from the author and date of the last comment (more recent the better). It is also a good practice to identify the common issues of the tool and make them available in the CWL wrapper’s description as given in the Bismark or Samtools Fastq tools.
Yes, workflows!
So, how to find “the magic of creation” in building a product using only off-the-shelf parts? It is a challenge to keep the same level of excitement as developing algorithms, but the wide usability of bioinformatic workflows is too vital to ignore. Obtained portability and reproducibility by using CWL and Docker is a must-have nowadays, especially with complex processing flows, many samples and requirement for harmonized results. In the end, a recent post in the bioinformatics community might just sum it all up: “Not every bioinformatician gets to go to the algorithm ball, somebody has to stay at home and write the workflows.” It looks like nowadays algorithm balls are almost empty since most of the bioinformaticians are sitting home writing the workflows.