GATK Best Practice: RNA-seq Variant Calling Workflow on the Seven Bridges Platform
RNA-seq variant calling
Whether or not variant calling should be performed on RNA-seq data and its possible benefits is a debatable topic. One thing is for certain, if you have already sequenced the transcriptome with the intent to analyze gene expression, but you are also interested in exploring variants existing in the same dataset, it is possible to do that. In order to do so, two significant considerations that need to be accounted for are the depth of coverage for lowly expressed genes and the inability to detect heterozygous variants due to allele-specific gene expression. If these two considerations don’t have you running for the hills, you’ll join a community of researchers who utilized RNA-seq variant calling as a standalone method or as a complement to whole exome sequencing (WES) to detect driver mutations in various types of tumors (1,2,3,4). Variant calling performed on RNA-seq from tumor samples offers a valuable addition to WES for several reasons. A major one is the ability of RNA-seq to identify new variants, which likely comes from higher sequencing coverage of significantly expressed genes. Oncogenes in cancers are likely to be highly expressed, and RNA-seq naturally provides better coverage of such genes than WES, and hence higher statistical confidence to detect variants. Also, a large number of highly expressed genes in cancer fall outside the boundaries of exon capture kits, thus not being detectable with WES. However, some RNA-seq variants could be considered questionable, as RNA-seq data is more prone to false-positive calls, in part due to errors during the RNA to cDNA conversion, mapping mismatches, RNA editing processes and the existence of splice sites and fusion genes.
To adequately address drawbacks of RNA-seq usage for calling variants and create a robust workflow that will produce high confidence results, the GATK team from the Broad Institute developed a Best Practices workflow for RNA-seq variant calling. The current implementation of this workflow can be used to identify germline variants but it is not recommended for somatic variant calling. Identification of germline variants could be a valuable asset when calling variants on tumor samples without corresponding matched normals.
CWL 1.0 implementation
Broad’s GATK team created this workflow and made it publically available in the Workflow Description Language (WDL) format. WDL is a convenient way to represent data processing workflows in a human-readable way. On the other hand, the Seven Bridges Platform uses the Common Workflow Language (CWL), a widely supported, open source specification for workflow descriptions. We used GATK’s WDL description of the Best Practices workflow for RNA-seq variant calling to create our implementation of this workflow in CWL format. While doing so, we managed not to make changes that will constitute a significant divergence from GATK’s Best Practice. We confirmed this by obtaining the same results after running the workflow with both WDL and CWL. Workflows created in CWL are highly portable and not restricted to the Seven Bridges execution environment. There is a wide variety of executors that support CWL and the same CWL document will run on a laptop, on high-performance computing clusters, on raw cloud infrastructure and on a variety of cloud platforms.
Overview of the workflow
GATK Best Practices RNA-seq workflow (Figure 1) starts from an unmapped BAM file containing raw sequencing reads. The sequencing reads are first mapped to the reference using STAR aligner (basic 2-pass method) to produce a file in BAM format sorted by coordinate. After marking and removing duplicates with Picard’s MarkDuplicate, StlitNCigarRieads splits reads with N characters in the CIGAR strings into separate reads and trims them to remove any overhangs into introns thus reducing the occurrence of artifacts in downstream processing steps. The remaining steps are similar to variant calling from DNA data, such as base recalibration performed with BQSR and variant calling performed with HaplotypeCaller. In the absence of the RNA-seq truth and training sets, it is necessary to apply hard filtering in the last step of the workflow, so VariantFiltration is used for this purpose. Because of HaplotypeCaller this pipeline should be used for germline variant calling.
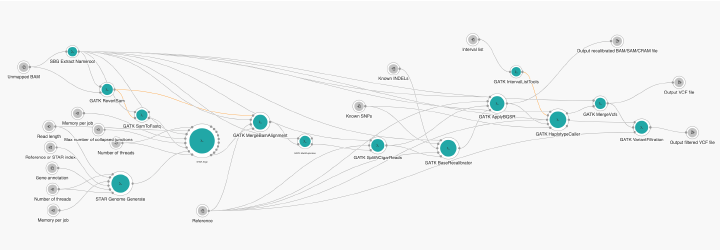
In the current implementation of the RNA-seq variant calling workflow, we used GATK 4.1.0.0 suite. For tools that are not within the GATK suite, we used adequate versions from WDL description for RNA-seq Best Practices.
Execution time and price
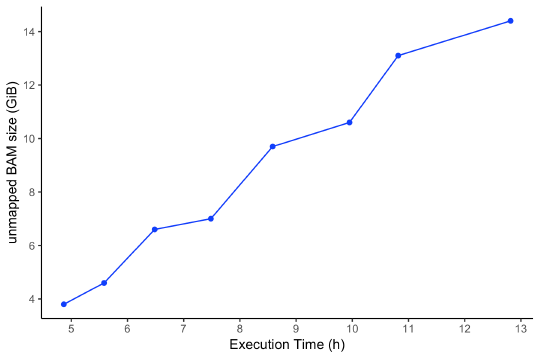
The runtime of this workflow on the Seven Bridges cloud infrastructure varies depending on the size of the input files. An unmapped BAM file containing raw sequencing reads is the factor that influences workflow execution time the most. In Figure 2, we represent the runtime execution (in hours) in comparison to the unmapped BAM file size (in GiB). The price of execution varies between $2.71 and $6.99 per sample, which could be significantly reduced (up to 75%) when using Amazon Web Services (AWS) spot instances.
To reduce the execution time of the workflow, you can change the default number of threads, used by STAR for generating index files and performing alignment to the reference. Also, you can set the number of parallel executions across chromosome intervals which is enabled for the HaplotypeCaller app using the scatter feature of CWL.
Running on the Seven Bridges Platform
Feel free to explore genetic variants in RNA-seq data using our BROAD Best Practice RNA-seq variant calling workflow. You could start off with examining the variants in some of the public datasets available from the NCI Genomic Data Commons (GDC), hosted on the Seven Bridges Platform. Also, we offer various solutions to easily upload your own data and run the analyses using our cloud infrastructure.
For more information on how to get started on the Seven Bridges Platform, contact us today.
References
- Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009 Oct;461(7265):809.
- Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, Gunawardana J, Jenkins C, Cochrane C, Ben-Neriah S, Tan K. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012 Mar 1;119(9):1963-71.
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012 Jun;486(7403):395.
- Coudray A, Battenhouse AM, Bucher P, Iyer VR. Detection and benchmarking of somatic mutations in cancer genomes using RNA-seq data. PeerJ. 2018 Jul 31;6:e5362.