Sequence Bloom Trees, Part I: Motivation and principles
The problem of subset membership testing
Modern bioinformatics involves a lot of searching datasets, like The Cancer Genome Atlas (TCGA), that contain data from many experiments. Wanting to do this efficiently raises not only data management problems but also algorithmic ones. Searching a dataset like TCGA in hopes of figuring out which experiments contain a given transcript suggests the problem of subset membership testing: given a sequence S and a domain D that is divided into subdomains D1, D2, …, Dn , in which subdomains {Di} does S appear?

Some fundamental techniques in algorithms suggest at least two approaches to this problem: first, we could find all matches of S in D and, using a correspondence between locations in D and D’s subdomains, find the relevant subdomain for each match. Finding matches of a string in a larger string (or set of strings) is a well-studied problem. So, for example, we could use a suitably optimized version of the Boyer–Moore algorithm, and accomplish this task in linear time. This speed is acceptable in many applications but often not in bioinformatics problems as datasets increase in size.
More promisingly, we could treat each subdomain Di as a separate domain, perform a fast set-inclusion test in each Di, and take the union of the subdomains corresponding to any positive tests. Set inclusion is another well-studied problem, and here, Bloom filters are a promising technique. By creating a Bloom filter for each of the subsets, we can search all the subdomains by doing constant work n times, so this will be much faster than the first approach. If n (the number of subdomains of D) is small, simply iterating through each subdomain will indeed often be the best approach. For larger n, however, this will generate work that we would like to eliminate. Consider the case where S does not appear in D at all: here the proposed method would perform n tests when all of them are destined to fail; we desire an algorithm that detects this failure at an earlier stage.
We have, then, two candidate algorithms, one of which is very inefficient and the other of which is moderately inefficient. Our difficulties arise because the subset membership problem occupies a middle ground between the pure set-membership problem that Bloom filters address and the matching-with-location problem that algorithms like Boyer-Moore address. We can solve the subset membership problem by treating it as a matching-with-location problem, but that requires working at a fine-grained level that generates vast extra work. Conversely, we can solve the problem by treating it as (several copies of) the set-membership problem, but doing this naively again generates extra work and fails to respect the structure of the problem.
Thankfully, some careful thought about the structure of Bloom filters, along with some basic techniques in graph traversal, allows us to do better than either of these approaches.
Sequence Bloom Trees offer a solution
For a while, we have known that the structure of Bloom filters allows us to build more complex and efficient data structures from them. Adina Crainiceanu’s work on Bloofi is an early example of this, and more recently, Solomon and Kingsford introduced Sequence Bloom Trees (SBTs), which apply this work to bioinformatics. SBTs retain the benefits of Bloom filters in a way that facilitates efficient searching through subsets; this improves on the approach where we blindly iterate through the subdomains. SBTs exploit a key insight about Bloom filters to allow them to respect the structure of the subset problem in a way that pure Bloom filters cannot.
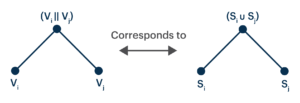
This key insight is that the bit vectors that comprise a Bloom filter have structure that give the bitwise OR operation a natural and useful interpretation: if Vi is a bit vector corresponding to the set Si and Vj is a bit vector corresponding to the set Sj, the bit vector (Vi || Vj) (where “||” denotes the bitwise OR operation) corresponds to the set (Si ∪ Sj).
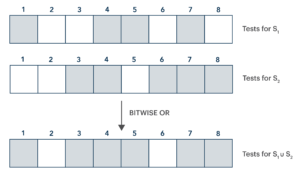
This correspondence follows immediately from the rule for constructing these bit vectors: if a bit is set to 1 in either Vi or Vj, that is because the relevant value is that of one of the hashes of some element of Si or of Sj—but this is just to say that it is the hash of some value in (Si ∪ Sj), and so would be set to 1 in the bit vector corresponding to that union.
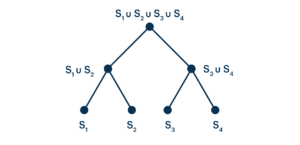
We can exploit this property to arrange bit vectors into trees.
Notice that nothing prevents us from iterating this procedure.
Given that we can arrange bit vectors into trees, we can use well-studied binary search techniques to organize and traverse these trees intelligently. More specifically, when we are looking for some sequence, we start by testing the root node of the tree for that sequence. A negative result means that the sequence does not appear in the union of the sets represented by the root’s daughter nodes—but this is just to say that a negative result means that the sequence does not appear anywhere in the data set.
A positive result, meanwhile, means that the sequence does appear somewhere in the data set. Thus, we need to continue the search, and proceed to the root’s daughter nodes. At each of those nodes, a negative result will mean that the sequence appears nowhere in the corresponding sub-tree, and a positive result will mean that it does. Repeating this process recursively, we will find exactly those leaf nodes in which the sequence appears. But to do this is just to solve the subset-inclusion problem.
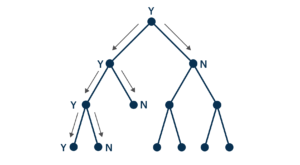
In the traversal depicted above, we can stop looking down through the tree every time we get a negative result from the node’s Bloom filter; it guarantees that the sequence in question appears nowhere in the relevant sub-tree. Of the 13 nodes, we avoid having to search 6 of them (the ones at the bottom right). The rarer a sequence is, and the more intelligently we arrange the tree, the more nodes we can skip in our search.
The correspondence between the bitwise AND of a Bloom filter and the union of the represented sets, then, is much more than just a formal observation; it is the key to a more efficient search when we have a lot of sets we want to search simultaneously. Thus SBTs give us the advantages of Bloom filters but also, by exploiting the structure of Bloom filters, allow a search that is logarithmic, rather than linear, in the number of subsets. For data sets comprised of hundreds of experiments (a common situation), this is a major improvement.
Looking ahead
These fundamental principles show how SBTs allow us to solve the subset membership testing problem more efficiently than other approaches. Our next posts in this series will extend these principles so that we can use SBTs as a practical bioinformatics tool. More generally, SBTs are a instructive case study in the ways in which bioinformatics is spurring us to push boundaries not only in data storage and transfer but also in data structures and algorithms.