Optimized Workflow for Bisulfite Sequencing Data Analysis
Epigenetics is an extra layer of information that is not encoded in the primary sequence of an organism’s DNA. While several mechanisms of epigenetic regulation exist, DNA methylation is one of the most commonly studied ones. Typically, the presence of methylated cytosines within a promoter region represses gene expression.
The extent and location of methylation can vary widely among species and amongst tissue types within a single species. By studying genome-wide methylation patterns in different cell types, species and during development, researchers gain insight into the mechanisms of epigenetic regulation in health and disease, as well as its impact on genome evolution.
Bisulfite Treatment Complicates Read Mapping
To determine methylation patterns, DNA is often treated with bisulfite to convert unmethylated cytosines to uracil. Sequencing libraries produced from this template incorporate thymine at the complementary position, allowing detection of the methylation event.
Alignment of libraries produced from bisulfite treatment of DNA is, however, more challenging both due to the low complexity of the underlying DNA regions and because the resulting sequence reads are no longer complementary to the reference genome.
Additionally, bisulfite-treated libraries can be of two distinct types: directional and non-directional. For directional libraries, adapters are designed in such a way that only original top and original bottom strand sequencing is enabled 1. Conversely, in non-directional libraries, all four DNA strands that result from bisulfite treatment and PCR amplification can be sequenced with roughly the same frequency 2,3. The subsequent alignment step may therefore require up to four different mapping processes for each read sequence, which greatly affects time and resources needed for analysis execution.
To mitigate this, Seven Bridges has implemented an optimized workflow for processing bisulfite sequencing data that takes advantage of elastic cloud resources to parallelize time-consuming steps. In short, researchers get their results faster without the need to install tools or dependencies.
Workflow for analyzing sequencing libraries from bisulfite treated samples
The workflow for processing libraries from bisulfite-treated samples (Figure 1) is based on the widely adopted Bismark4 toolkit which deploys three-letter mapping algorithm and uses Bowtie as an internal aligner.
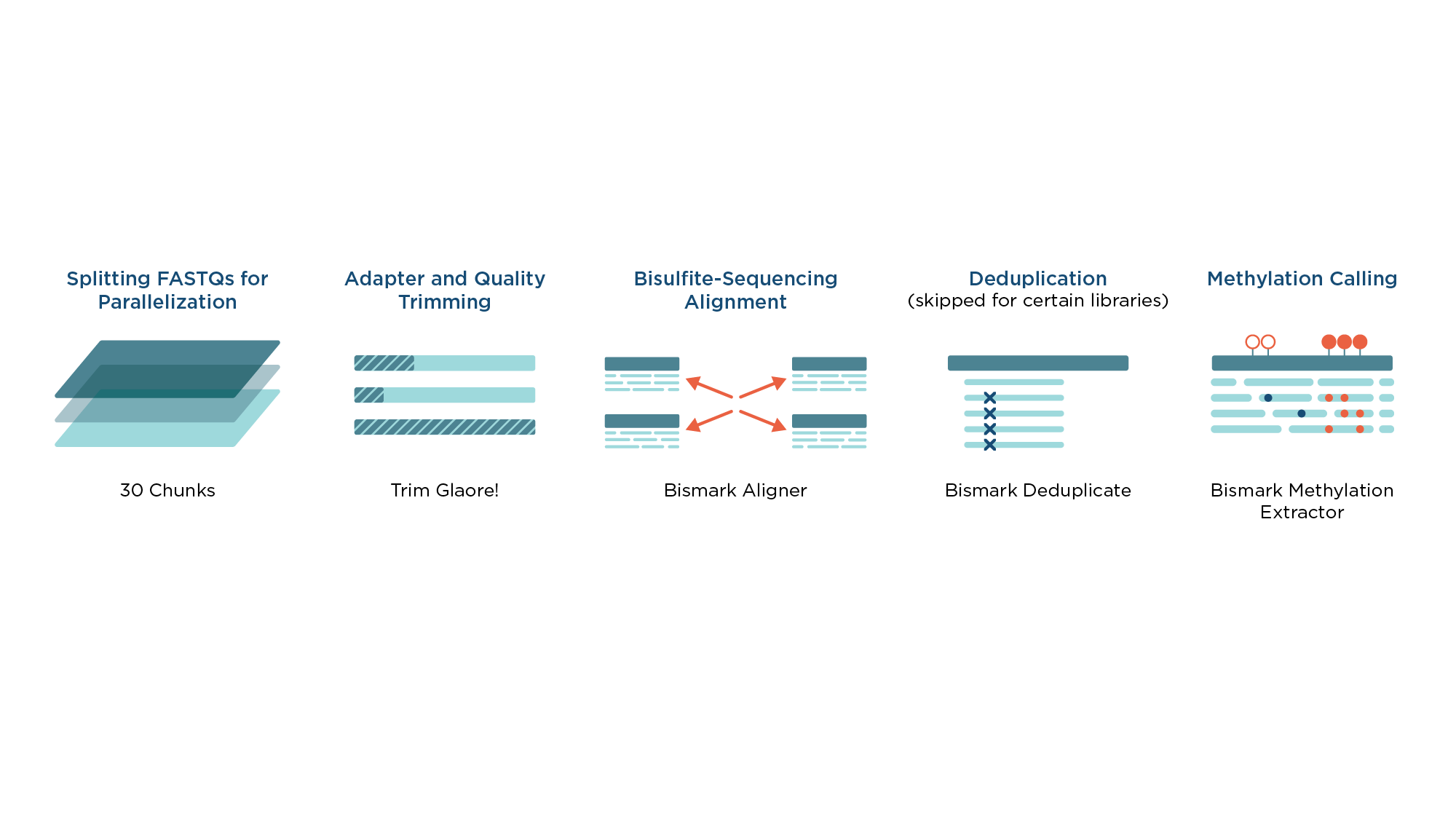
Prior to alignment, reads are processed by TrimGalore! to remove low-quality sequence and FastQC Analysis is performed before and after trimming.
The trimmed reads are mapped with the Bismark aligner and duplicate reads are removed with Bismark Deduplicate. Bismark Methylation Extractor provides final methylation calls and methylation percentages per each CpG site.
Speeding up the workflow with elastic resources
To speed up the workflow, the input FASTQ files are split into multiple files and Bismark alignment is executed for each batch on several different EC2 instances. Simultaneous alignment of each group of sequenced reads was performed by utilizing the multi-instance scheduling feature, which allows fully automated allocation of multiple machines.
Furthermore, the methylation extraction step is parallelized by individual chromosomes. First, read alignments obtained as a result of Bismark execution are merged according to the reference contigs from which they originate, which results in 93 BAM files in case of UCSC hg19 reference assembly. Then, Bismark Methylation Extractor job is generated for each contig and these runs are scheduled on multiple machines.
Interestingly, by increasing the number of EC2 instances used, the execution time is significantly decreased so that the cost also drops despite the fact that multiple machines are allocated throughout the whole analysis.
More specifically, multi-instance scheduling was set to allocate a maximum of 6 instances, all of which are c4.8xlarge (36 CPUs, 60GB of memory). This exact number is a result of testing with several different settings for the number of instances. For lower number of instances, execution was slower without notable savings in terms of the cost and for higher numbers, there wasn’t a significant speed-up considering the increasing costs.
Input reads are split into 30 different (pairs of) files and passed to TrimGalore! for parallel execution. Considering the fact that there are FastQC processes running simultaneously with trimming, the full instance capacity (36 CPUs) wasn’t utilized only for TrimGalore!. In order to have only one instance allocated for both TrimGalore! and FastQC, FASTQs are split so that 30 CPUs are consumed by trimming and the rest is available for FastQC processes. In case of a higher number of FASTQ chunks, more than one instance would be allocated automatically, which is needless considering the low complexity of those executions. Subsequently, Bismark alignment and Bismark Methylation Extractor are run each in parallel — using 6 instances at the most.
Performance statistics
As mentioned earlier, alignment of bisulfite-treated reads poses a considerable computational challenge. Performance of Bismark aligner highly correlates with sequencing library (either directional or non-directional), the size of the reference genome file, and the number of multi-cores assigned to Bismark. Therefore, we tested both sequential and optimized version of the workflow with many data sets, two of which are chosen as an example — WGBS data derived from human colon tumor sample5 and oxBS-seq together with BS-seq data derived from spiked-in human sample6 (Table 1).
| Sample | Sample information | Indexed reference genome |
| BS-seq (SRR949210) |
|
|
| oxBS-seq + BS-seq (SRR2074693) |
|
|
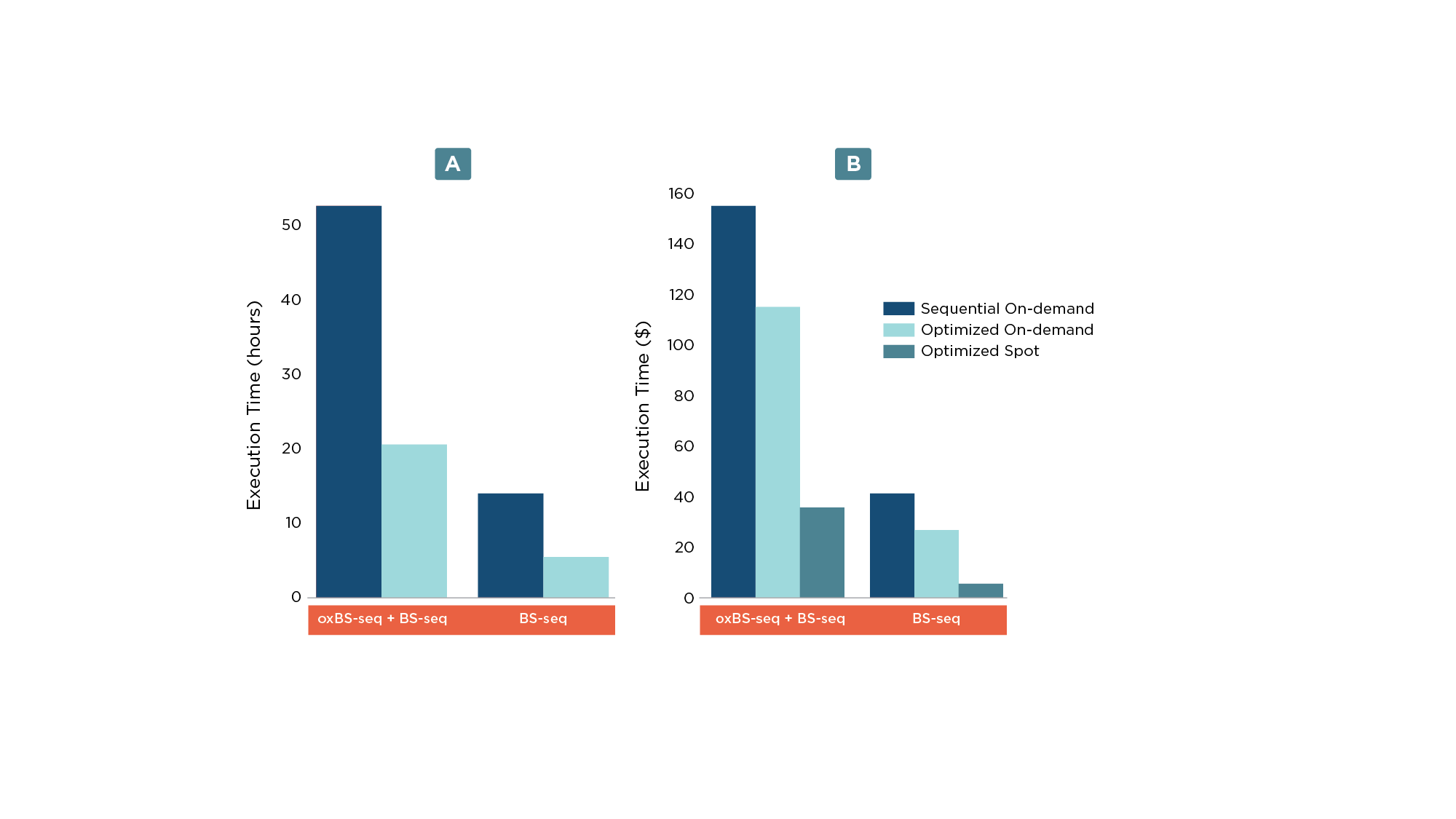
Utilizing multi-instance scheduling enabled more than a two-fold decrease in the execution time (Figure 2a) resulting in a cost reduction of ~26% (Figure 2b).
Reducing the cost with spot instances
In addition to running an optimized workflow, execution cost can be further reduced by using Amazon EC2 Spot instances (Figure 2b). Amazon EC2 Spot instances are spare compute capacity and by bidding on it, Seven Bridges allows analysis cost optimization and savings of up to 90% compared to On-Demand prices. However, if the market price gets higher than the bidding price, the Spot instance gets terminated and the Seven Bridges Platform restarts unfinished jobs on an On-Demand instance.
This can result in increased execution costs and analysis duration. For this reason, it is crucial to plan the work ahead and to take into account all the factors that may affect Spot instance prices, e.g. instance type, current pricing trend, number of executions etc. Additional information related to Spot prices history can be found on Spot Instance Advisor.
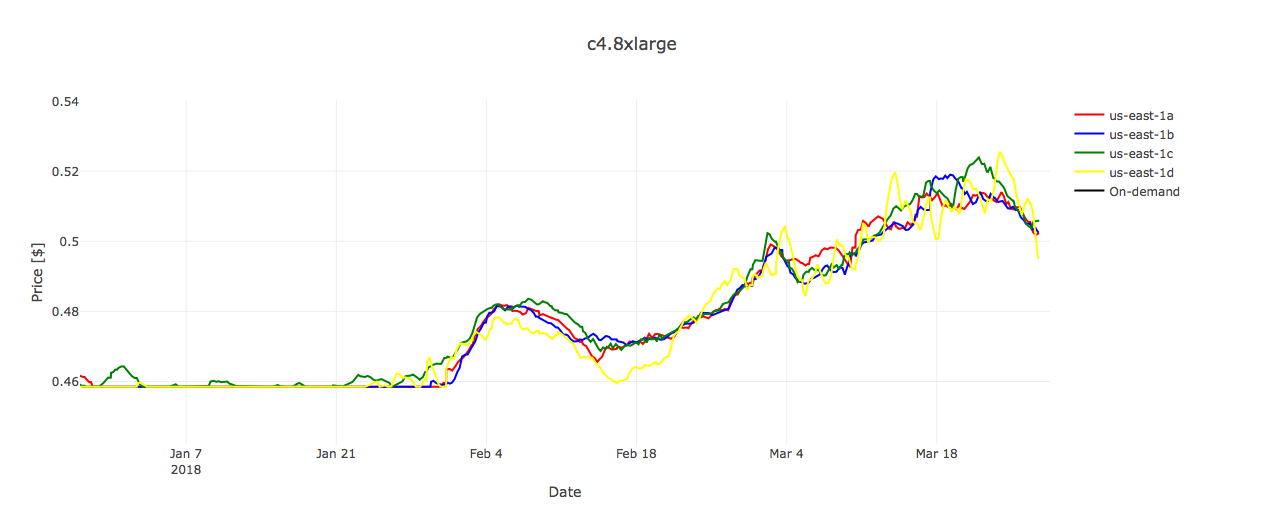
Regarding the Bismark workflow, it is important to note that running only one analysis requires allocation of 6 instances, which inevitably increases the demand of Spot instances and, in case of several parallel executions, can increase the chances for market price to exceed the On-Demand price.
However, if we take a look at the prices diagram for c4.8xlarge (Figure 3), we can see that the Spot price was always substantially below the fixed On-Demand price during the period of 90 days.
Thus, if tasks are carefully executed (e.g. in smaller batches rather than all at once), it is possible that everything is going to play out well — without any surprises and with quite some savings on the billing account.
References
- Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315-322. doi:10.1038/nature08514.
- Cokus SJ, Feng S, Zhang X, et al. Shotgun bisulfite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452(7184):215-219. doi:10.1038/nature06745.
- Popp C, Dean W, Feng S, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by Aid deficiency. Nature. 2010;463(7284):1101-1105. doi:10.1038/nature08829.
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571-1572. doi:10.1093/bioinformatics/btr167.
- Ziller MJ, Gu H, Müller F, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500(7463):477-481. doi:10.1038/nature12433.
- Li X, Liu Y, Salz T, Hansen KD, Feinberg A. Whole-genome analysis of the methylome and hydroxymethylome in normal and malignant lung and liver. Genome Research. 2016;26(12):1730-1741. doi:10.1101/gr.211854.116.