Identifying viral sequences in TCGA data using Kraken and Centrifuge
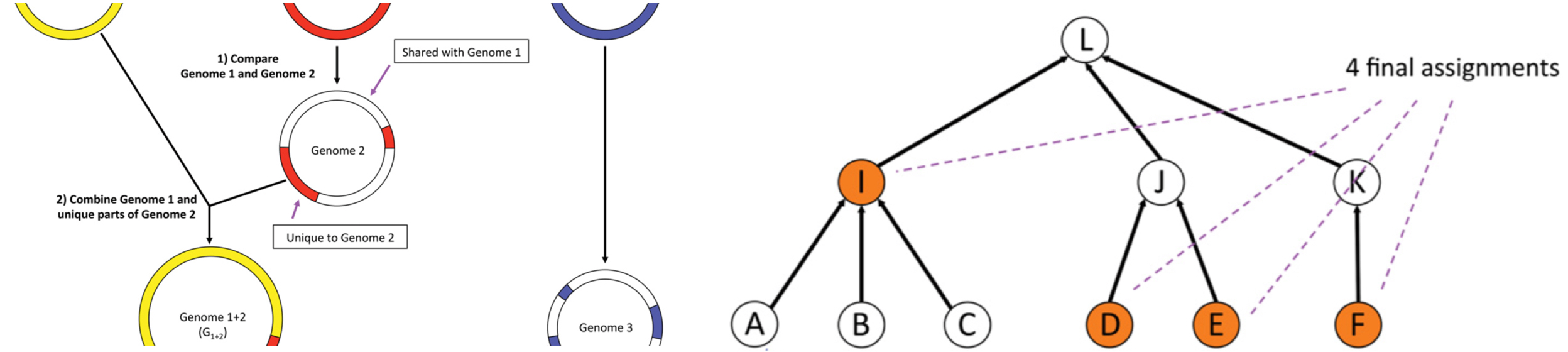
Image adapted from Kim et al. Genome Res. 26, 1721–1729 (2016).
Next-Generation Sequencing has opened up the field of metagenomics. In metagenomic studies, a sample often contains a complex ecosystem of different microorganisms. The key challenge in these experiments is disentangling the identities of unknown species from millions of sequencing reads. Bioinformatic tools for metagenomics are designed to systematically look through sequence information and return a list of species and their relative abundance.
Metagenomic approaches have been used to great effect to understand the diversity of bacteria in the human intestine, and to characterize the microenvironment in samples ranging from seawater to cell phones.1–3
In this blog post, we’ll discuss two bioinformatic tools for metagenomics, and test their performance on the Seven Bridges Cancer Genomics Cloud (CGC) by identifying viral sequences in cervical cancer samples from The Cancer Genome Atlas (TCGA).
About the tools
Kraken and Centrifuge are two powerful metagenomics tools developed by the Salzberg lab at Johns Hopkins Center for Computational Biology. Both tools are highly accurate and are orders of magnitude faster than BLAST-based algorithms.
With earlier tools, researchers were faced with a trade off between reasonable analysis time frame and accuracy. BLAST-based tools, while accurate, are very slow and computationally expensive. To perform more sophisticated metagenomic experiments, researchers need tools that can quickly and accurately derive useful information from a tangled web of sequencing reads.
Kraken and Centrifuge both use a reference database to classify sequences to their taxonomic ranks. Kraken searches for exact k-mer matches in a given sequence against the database to identify a taxonomic label for the sequence. It relies on a precomputed database of k-mers (substrings of fixed length k) and the lowest common ancestors of the reference genomes containing those k-mers. Kraken is able to classify 4.1 million 100-base-pair reads per minute at accuracy comparable to the fastest BLAST program. 4
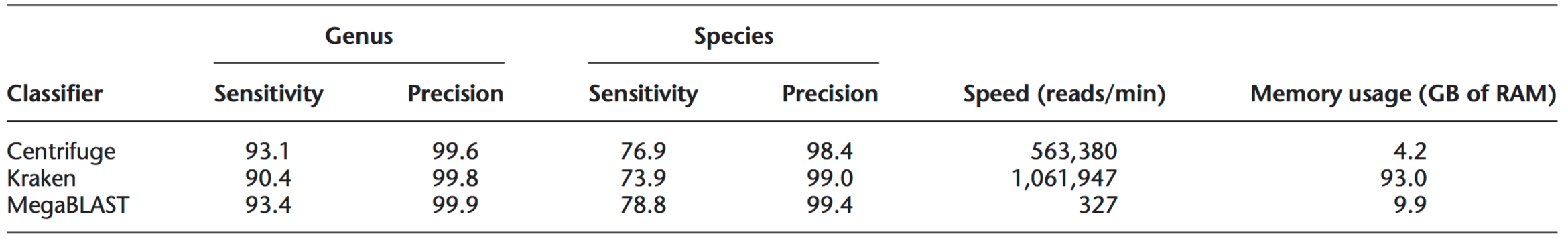
Table 1. Classification sensitivity and precision for Centrifuge, Kraken, and MegaBLAST using simulated reads. In Centrifuge, only uniquely classified reads were used to compute accuracy. To measure speed, 10 million reads were used for Centrifuge and Kraken and 100,000 reads were used for MegaBLAST. All programs were run on a Linux System with 1TB of RAM using one CPU (2.1 GHz Intel Xeon). Table reproduced from Kim et al. Genome Biology 2016.5
A drawback of Kraken is its substantial memory requirement. For a database of 4,278 prokaryotic genomes, Kraken requires 93GB of memory, effectively restricting its use to high-performance clusters.
Centrifuge was developed to address this drawback. By exchanging k-mer based indexing with a strategy based on the Burrows-Wheeler transform and Ferragina-Manzini index, Centrifuge significantly reduces the storage space for the indexed reference genomes, while still allowing quick exact search of k-mers for a given sequence.5
The reduced storage space translates to a lower memory footprint in Centrifuge. A centrifuge index of the full NCBI non-redundant nucleotide sequence database requires only 69 GB, and includes full taxonomical mapping of the sequences in the sample.
Centrifuge maintains high levels of accuracy and can be effectively run on a personal computer. Unlike Kraken, Centrifuge can assign a sequence to multiple taxonomic categories, allowing more specificity at the genus and species level (Table 1).
Experimental design
To better compare the performance of these tools, we designed a simple experiment to quantify viral transcripts in data from TCGA.
TCGA hosts more than 2.5 PB of high-quality molecular data from 33 types of cancer. We used the CGC to pull RNA-seq data from 304 cervical tumors sequenced as part of TCGA.
Given that human papillomavirus (HPV) is implicated in more than 70% of cervical cancers,6 we wanted to see if we could detect HPV sequence within TCGA’s cervical cancer RNA-seq datasets.
We found extensive viral transcription from the pathogenic HPV strains HPV16 and HPV18 in these cancers. Let’s look in depth at how we used CGC to run this experiment.
Gathering the data: samples
We began by collecting the RNA-seq data from all cervical cancer tumor samples present in TCGA. This is easy to do using the CGC Data Browser (Figure 1). First, we selected the cancer sample classes Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma and set the sample type to Primary Tumor. In the File block, we set Data Format to TARGZ (as TCGA hosts raw data as TAR.GZ bundles) and picked RNA-seq as our experimental strategy. This search gave us 304 TAR.GZ bundles to work with.
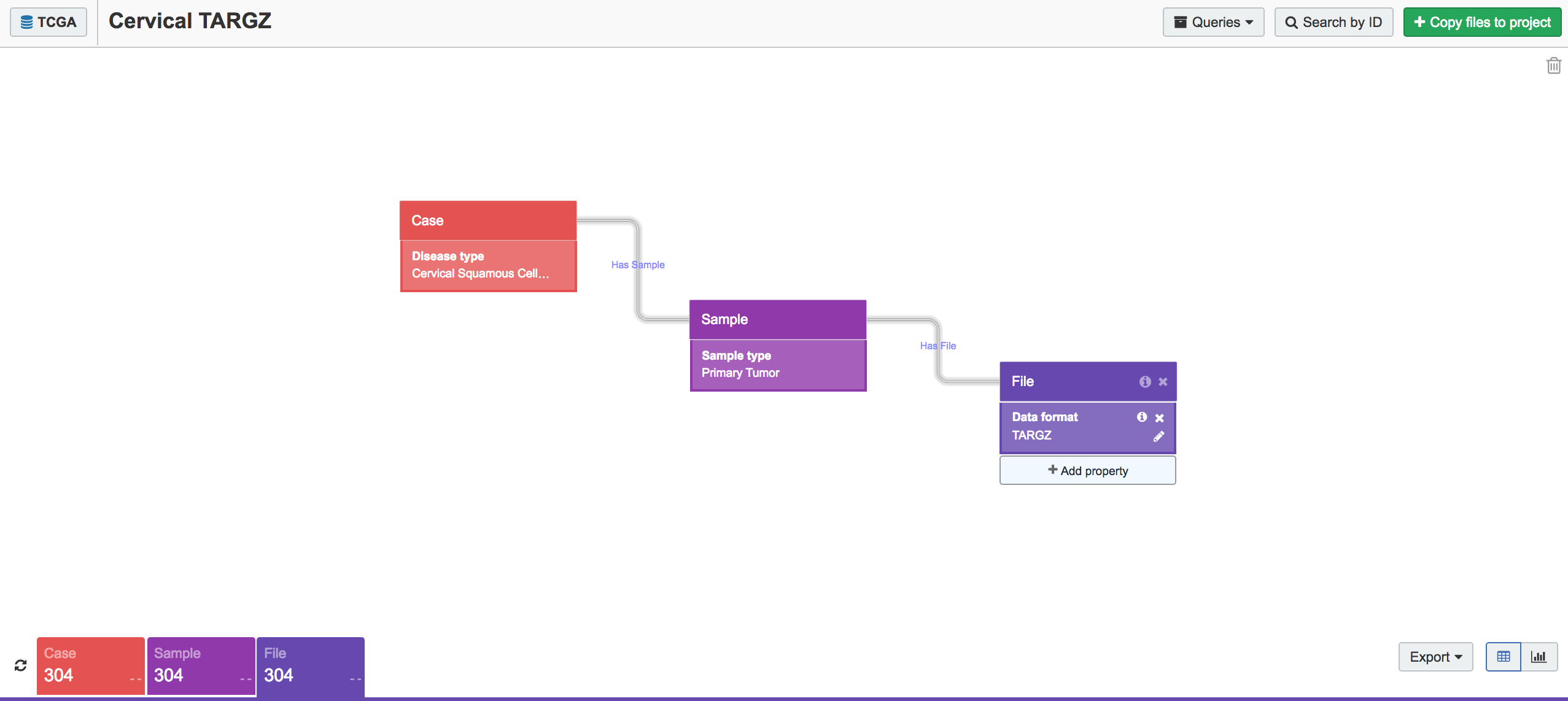
Figure 1. Data Browser query for raw RNA-seq sequences from cervical cancer primary tumors. The Data Browser makes it easy to identify and export samples of interest from within TCGA.
Building a custom reference index
Kraken and Centrifuge each require a reference genome database, which is used to query a given sample and to create indices for Kraken and Centrifuge. Centrifuge has a collection of pre-built indices available at their website, and Kraken also has a pre-built database of bacterial, archaeal, and viral genomes.
As there was no pre-built, viral-only index available, we chose to build the viral indices from scratch on the CGC. We first downloaded all 8,953 NCBI-cataloged viral sequences and then added 183 HPV reference sequences. In total, we had 9,136 viral reference genomes ready for comparison with the tumor sequences.
To build the Kraken index, we first converted the NCBI accession IDs from the viral reference sequences and then mapped them to their GI IDs.
With cervical cancer data and custom indices in place, we were ready to run our analysis.
Kraken
As the cervical cancer samples’ raw sequences are hosted by TCGA in tar bundles, we first used Seven Bridges’ Unpack FASTQs app to unpack the tar bundle and obtain raw sequences. Using the CGC workflow editor, we created a workflow chaining Unpack FASTQs and Kraken together so that the tar bundle can be passed directly through to the Kraken classifier app (Figure 2).
Since we wanted to process multiple samples from the cervical cancer cohort, we used the CGC’s scatter function to process our samples in parallel on a single task.
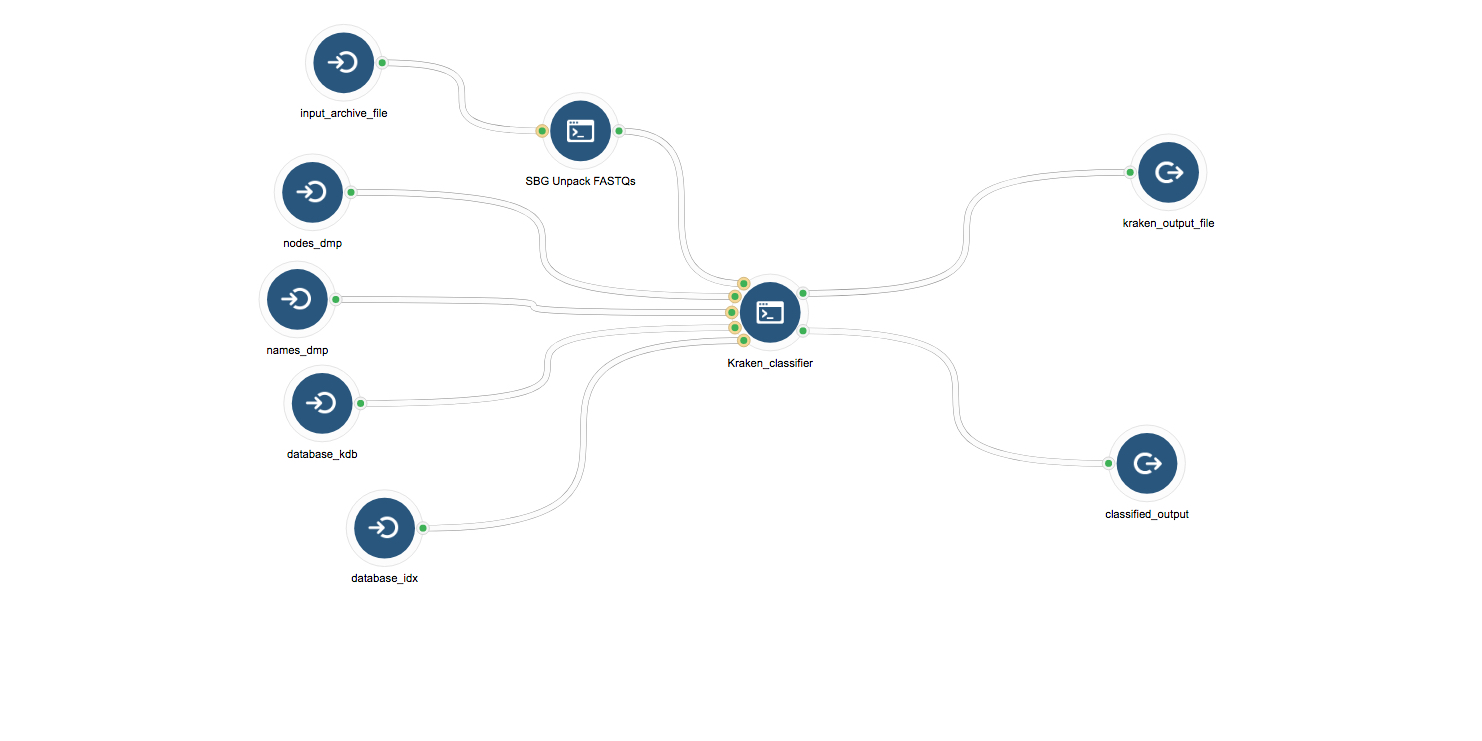 Figure 2: Kraken workflow. Kraken requires two types of inputs: a fastq file containing raw sequences from the sample, and reference index files corresponding to the reference genomes we want to search for. Two output files are produced: a summary of the genomes identified in the sample, and a classification of each read’s assignment to a reference genome.
Figure 2: Kraken workflow. Kraken requires two types of inputs: a fastq file containing raw sequences from the sample, and reference index files corresponding to the reference genomes we want to search for. Two output files are produced: a summary of the genomes identified in the sample, and a classification of each read’s assignment to a reference genome.
Runtime and execution analysis
We ran Kraken on 56 cervical cancer samples in a single task on the CGC (Figure 2). Each task acquires 4 c4.2xlarge AWS instances and analyzes varying number of samples. The number of samples in a single task was chosen so that the total disk space of all input files did not exceed 260 GB. To run all 304 samples, we subdivided our analysis into eight tasks.
As Kraken requires large memory, we conservatively ran only a single sample on a single c4.2xlarge instance at a time. The unpacking of tar bundles does not have such memory bottlenecks, which meant we could decompress multiple samples in parallel on a single instance.
Our analysis completed in 44 hours and cost $80. Kraken detected HPV transcripts in 241 of 304 TCGA cervical cancer samples.
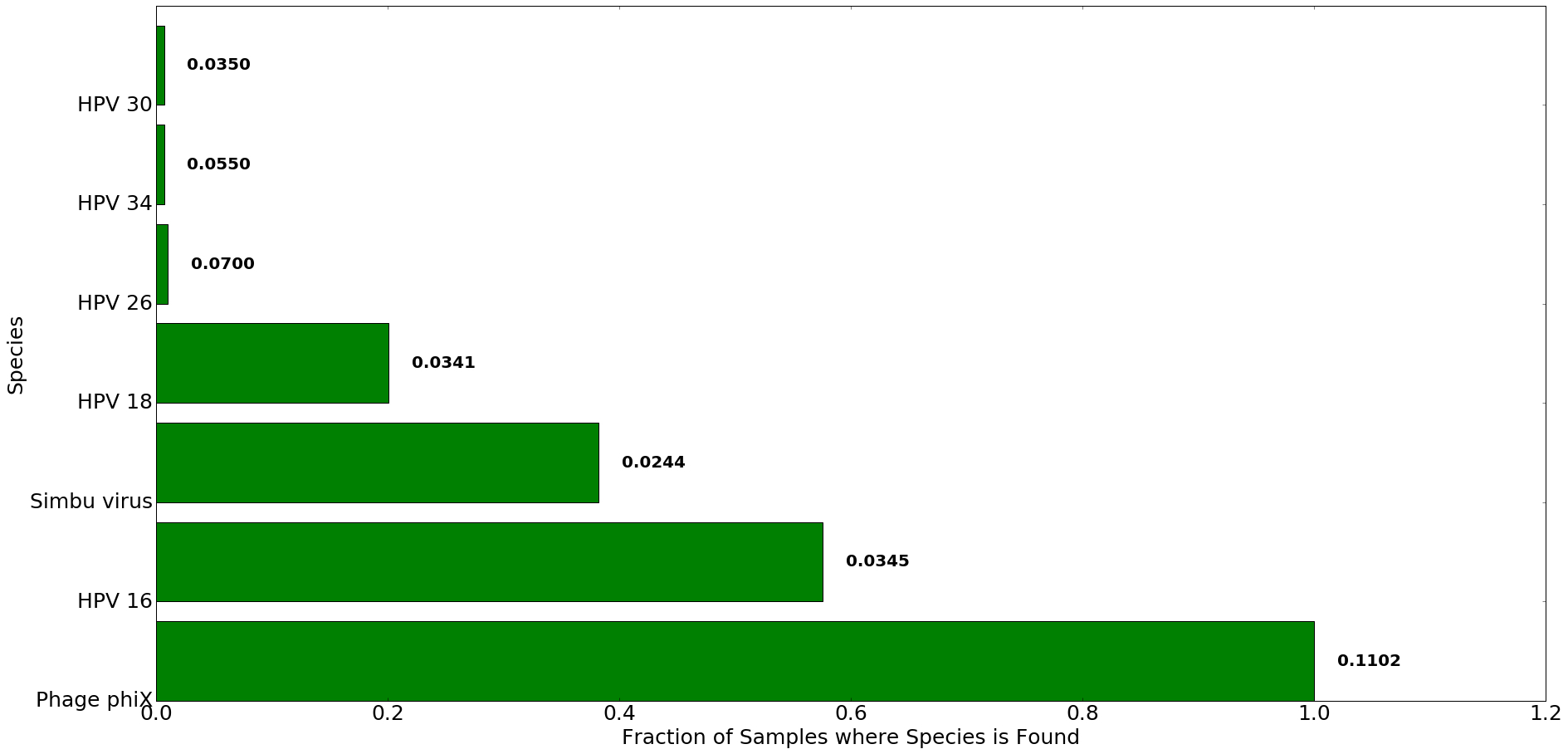
Figure 3: Top species detected in patients with cervical cancer using Kraken. The x-axis denotes the fraction of all samples in which species listed on y-axis was found. The number to the right of each bar indicates the average proportion of reads that mapped to each species in Kraken across all samples. Only species where average proportion was greater than 0.02 are were used for analysis and comparison.
Centrifuge
Next, we ran the experiment again, this time using Centrifuge. For this analysis, we used the CGC’s scatter functionality to process multiple samples in parallel. Additionally, we used the stream file read functionality to pull tar bundles directly from an S3 storage Amazon bucket, and incorporated stream read for fastq files directly in the command line for Centrifuge app, allowing us to convert the tar bundle of samples to fastq files on the fly without copying them onto the instance (Figure 4).
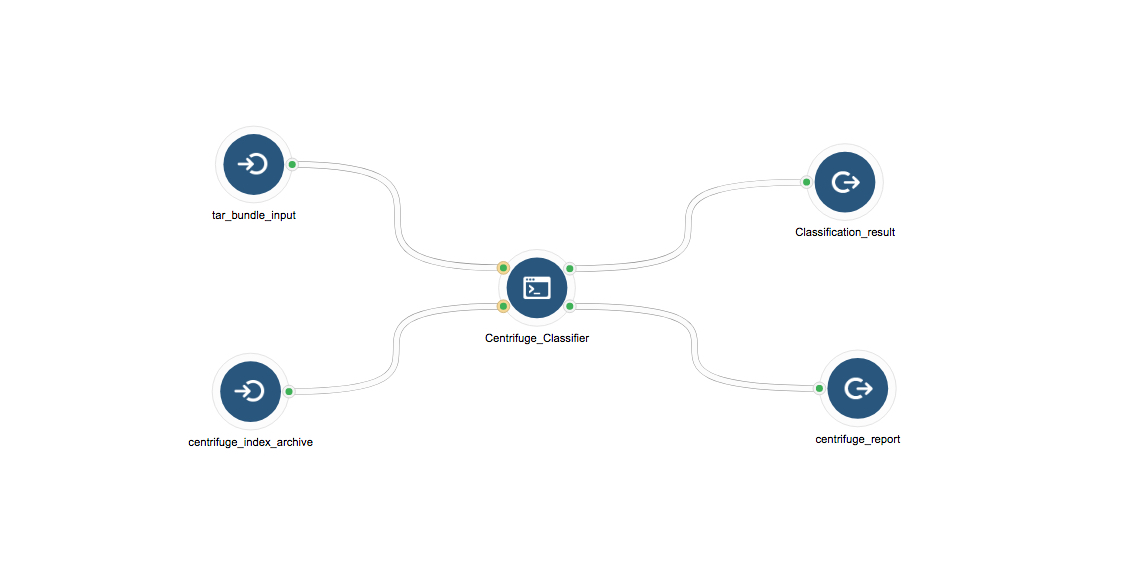 Figure 4: Centrifuge Classification Workflow. Centrifuge requires two inputs: the raw sequences from a sample as a fastq or fasta file, and the Centrifuge reference archive that contains reference indices. Centrifuge outputs two files containing results on a per read basis and giving summary statistics.
Figure 4: Centrifuge Classification Workflow. Centrifuge requires two inputs: the raw sequences from a sample as a fastq or fasta file, and the Centrifuge reference archive that contains reference indices. Centrifuge outputs two files containing results on a per read basis and giving summary statistics.
Centrifuge required a single task with four c4.2xlarge AWS instances to process all 304 cervical cancer samples. As Centrifuge has a low memory footprint, we could utilize the scatter functionality to schedule 8 samples in parallel on a single instance (Figure 5). The analysis ran for 8 hours and cost $14. Centrifuge identified HPV transcripts in 215 of 304 TCGA samples (Figure 6).
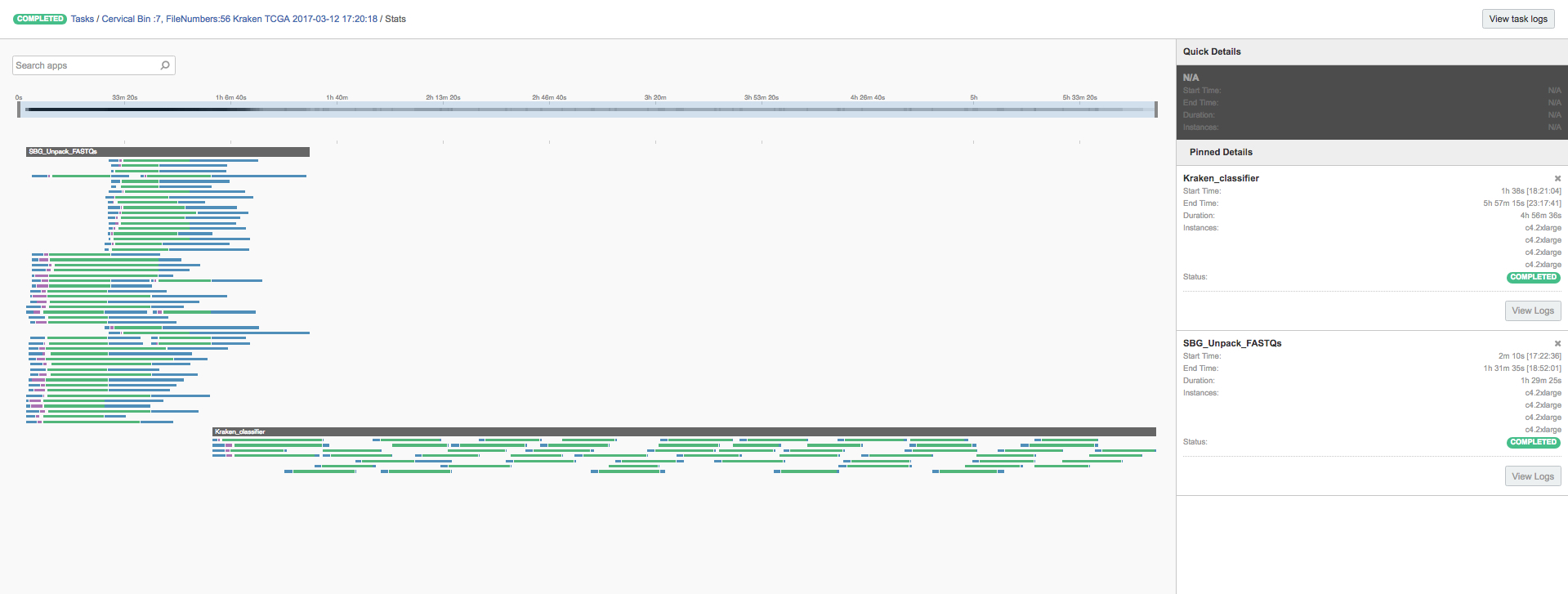
Figure 5: Centrifuge run over 304 cervical cancer samples from TCGA scattered over 4 AWS instances.
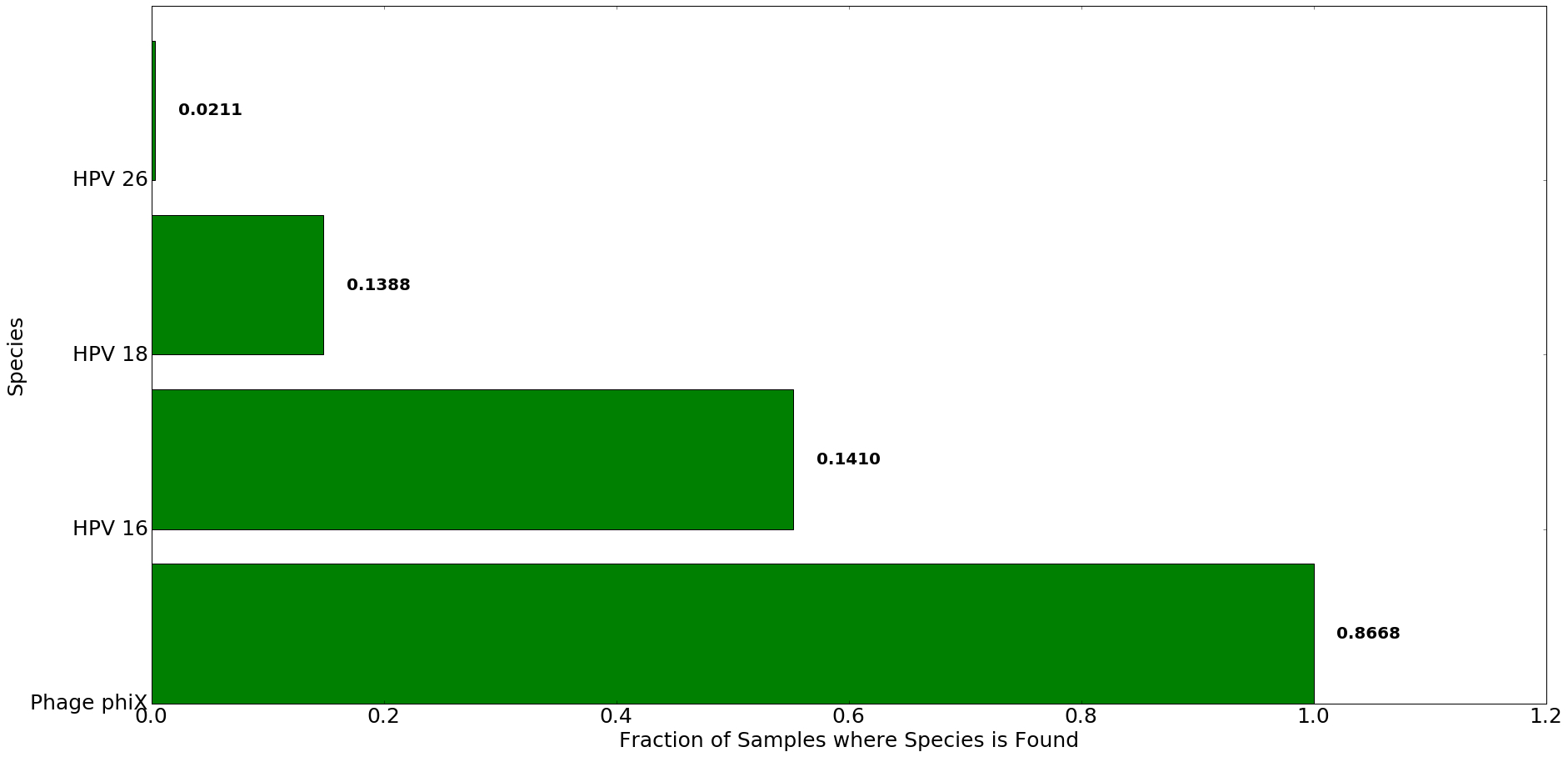
Figure 6: Most abundant species detected in patients with cervical cancer using Centrifuge. The x-axis denotes the fraction of all samples in which species listed on y-axis was found. The number to the right of each bar indicates the average abundance of each species in Centrifuge across all samples. A 0.02 abundance threshold was used.
Results & discussion
Overall, Kraken and Centrifuge produced similar results. Kraken and Centrifuge detected HPV16 and HPV18 in a majority of samples. It would appear that the connection between these pathogenic HPV strains and cervical cancer is recapitulated well in the context of TCGA.
Kraken detects the presence of transcripts from HPV 16 strain in 175 samples (57%), while Centrifuge detects it in 168 samples (55%). Another common strain associated with Cervical Cancer samples is HPV 18, which was found by Kraken and Centrifuge in 61 and 45 samples, respectively. Our analysis also pulled out two less common HPV strains, HPV 34 and HPV 26.
All tested samples contained sequences from phiX bacteriophage. This is expected, as phiX is a common control library used in illumina sequencing.
Kraken also identified Simbu virus sequences in about 40% of samples. It’s worth noting that only 2.44% of reads per sample were mapped as originating from Simbu virus. While it’s possible that Simbu sequences do indeed exist in our samples, the fact that they were identified by Kraken and not Centrifuge means this result is likely to be an artifact of Kraken’s indexing process. Centrifuge addresses this issue by allowing a given sequence in the index to be assigned to multiple species. This is a more nuanced approach to sequence homology within the index.
As a control, we also ran Centrifuge on RNA-seq data from glioblastomas from TCGA . Although glioblastomas can result from a variety of causes, we expected that HPV16 and HPV18 sequences would be less common here than in cervical cancers.
As expected, Centrifuge did not detect HPV transcripts in these control samples.
Summary
Our goal was to evaluate the performance of Centrifuge and Kraken for detecting viral transcripts across a cancer cohort. Centrifuge and Kraken produce very similar results and consistently detect HPV strains in cervical cancer. However, Centrifuge is significantly cheaper and faster than Kraken. Our Centrifuge run cost $14, while the Kraken run cost $80 on the same computation architecture. Furthermore, Kraken took roughly 44 hours to finish all analysis, while Centrifuge processed all samples in about 8 hours at a fraction of the price. We therefore recommend using Centrifuge for efficient memory, time and cost usage in metagenomics experiments.
If you are attending BioIT World 2017, stop by and see us! We plan to present this work alongside several other projects. Seven Bridges is located at booth #432.
- Breitbart, M. et al. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. U. S. A. 99, 14250–14255 (2002).
- Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010).
- Bouslimani, A. et al. Lifestyle chemistries from phones for individual profiling. Proc. Natl. Acad. Sci. U. S. A. 113, E7645–E7654 (2016).
- Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014).
- Kim, D., Song, L., Breitwieser, F. P. & Salzberg, S. L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729 (2016).
- CDC – How Many Cancers Are Linked with HPV Each Year? Available at: https://www.cdc.gov/cancer/hpv/statistics/cases.htm. (Accessed: 17th May 2017)

This work is licensed under a Creative Commons Attribution 4.0 International License.