GATK Best Practices Spotlight: The GATK Somatic Create Mutect2 Panel of Normals 4.1.9.0 workflow
The GATK Somatic Create Mutect2 Panel of Normals (PON) workflow takes multiple normal sample callsets produced by GATK Somatic SNVs and INDELs 4.1.9.0 workflow tumor-only mode (although it is called tumor-only, normal samples are given as the input) and collates sites present in two or more samples into a sites-only VCF. The PON workflow creates a panel of normals (germline and artifactual sites) for use in other GATK workflows. This tool and other Mutect2 series tools (see the original publication on BioRxiv) is an improvement upon the original “MuTect” tool created by Cibulskis et al, detailed in the Nature Biotechnology article publication here in 2013. Both the PON workflow and the SNVs and INDEL workflow are composed in reference to their official GATK’s WDL versions counterparts.
For more information on the associated GATK Somatic SNVs and INDELs 4.1.9.0 workflow, see our blog post here.
Using this workflow for your research
The PON workflow (Figure 1, below) captures common artifactual and germline variant sites and outputs them in the form of a VCF file. The main Mutect2 workflow then uses that file to filter variants at the site-level.
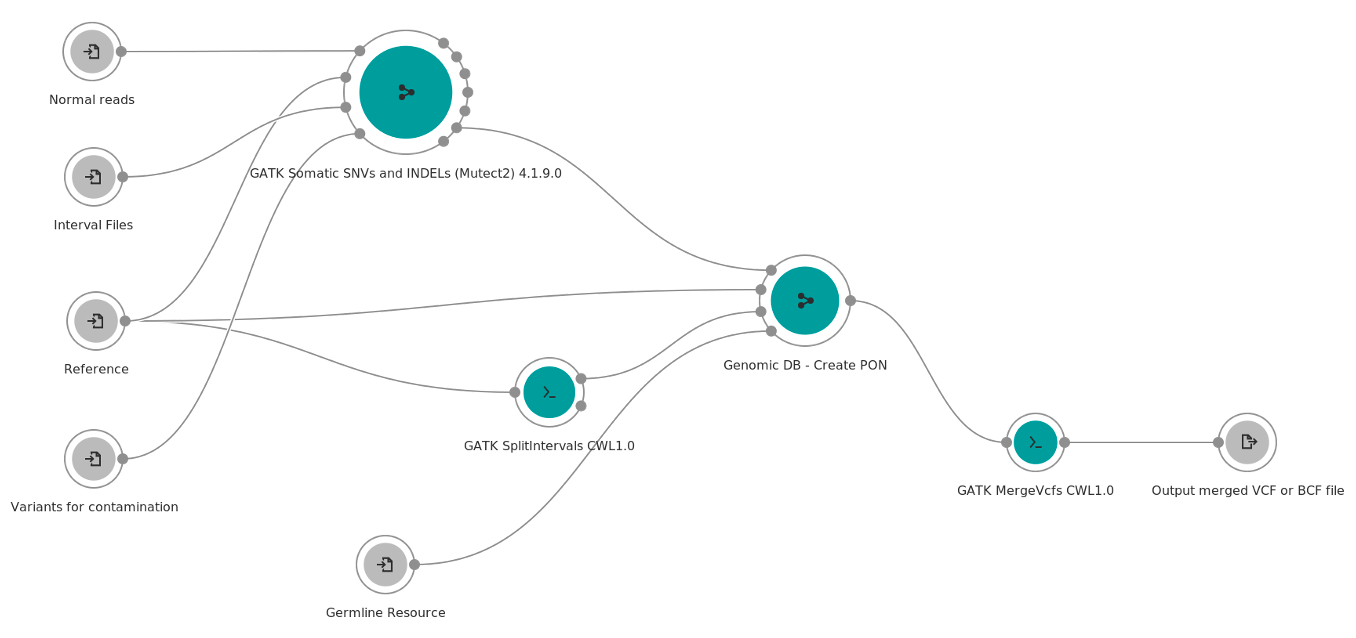
Figure 1: Overview of the GATK Somatic Create Mutect2 Panel of Normals workflow available on the Seven Bridges platforms.
As shown in Figure 1 above, the main workflow steps are:
- The first step of this workflow is to run the GATK Somatic SNVs and INDELs 4.1.9.0 in tumor-only mode for each normal sample.
- The next step is creating a GenomicDB from the normal calls.
- Third, the GATK Genomics DB Import tool is used to combine multiple single-sample GVCF/VCF files before joint genotyping, on the specified genomic interval.
- Finally, the output of the GATK Genomics DB Import is then used by the Create Somatic Panel Of Normals
The GATK Somatic SNVs and INDELs 4.1.9.0 workflow inside the GATK Somatic Create Mutect2 Panel of Normals 4.1.9.0 is parallelized (scattered) by Normal reads. The Genomics DB Import tool and Create Somatic Panel Of Normals tool are both wrapped as an inner workflow inside the main workflow so that they can be parallelized together.
Important notes for using this workflow
- The BAM inputs should be sorted and indexed. This can be done by using Bamtools, Picard, or Sambamba tools which are all available in the Public Apps Gallery.
- Reference FASTA and index require FAI and DICT index files. These files can be generated by using the SBG FASTA Indices
- Lastly, Germline resource (gnomAD) file and its index – database of known germline variants, (see The Broad Institute’s Site – gnomad downloads) is an optional but recommended input.
Performance Benchmarking
At Seven Bridges, our Bioinformatics Team performs benchmarking on many of our tools and workflows, in order to give users a better understanding on execution time and cost. In the table below, we represent the runtime execution cost in comparison to the unmapped BAM file size. The runtime of this workflow on the Seven Bridges cloud infrastructure varies proportionally with the size of the input files. The size of an unmapped BAM file containing raw sequencing reads has the largest influence on workflow execution time.
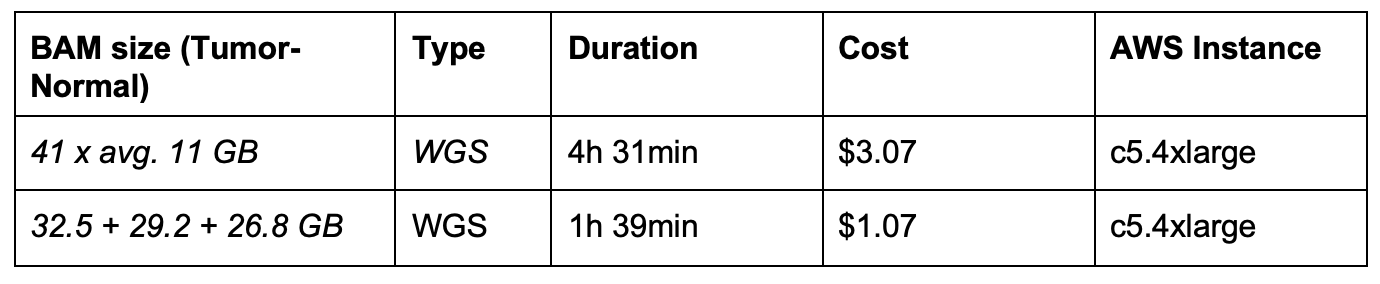
The price of execution for this workflow varying with input size is displayed in the table above. These prices could be significantly reduced (up to 75%) when using Amazon Web Services (AWS) spot instances. To learn more about spot instances, visit our Knowledge Center.
Running on the Seven Bridges platforms
A good starting point for running this workflow on the Seven Bridges could be comparing your patient data of interest with normal (without-tumor) blood samples from healthy individuals on The Cancer Genome Atlas (TCGA) dataset. In addition to our hosted datasets, we also feature a variety of methods to easily upload your own data to the Seven Bridges environment, and to run the subsequent analyses using our cloud infrastructure. For more information on how to get started on the Seven Bridges platforms, contact us today.