A first look at GATK4 on the Seven Bridges Platform
GATK4 is moving to a fully open source license
One of the big take-away messages from the Bio-It World Conference this year was the Broad Institute’s announcement that they plan to fully open source their GATK4 software. By transitioning to a BSD 3-Clause licence, GATK4 becomes fully open for commercial use without a separate commercial licence, which should particularly benefit researchers in start-ups, biotechs and pharma who make use of genomic data in their research activities.
Broad Institute makes genomic analytics software open source https://t.co/lNoQOY6lQb via @HealthITNews
— Broad Institute (@broadinstitute) May 26, 2017
The first version of the the Genome Analysis Toolkit (GATK) was published in 2010. Since then, GATK Best Practices have become a community standard for genotyping and variant calling in Next-Generation Sequencing (NGS) data. Despite the availability of multiple competing methods, our GATK-based whole-genome and whole-exome analysis workflows remain among the most popular for users of our Platform and other research environments.
Earlier this month, we released our first GATK4 workflow (Whole Genome Pipeline – BWA + GATK 4.0) across all the Seven Bridges environments. This workflow is based on the beta version of GATK4 currently available on the GATK github repository (the full release is anticipated later in 2017).
From conversations at conferences and with customers we understand that bioinformaticians are keen to access the latest beta version of the GATK4 workflow for variant calling using local and cloud computation resources. With the full general release of GATK4 pending, we take the opportunity to show some of our work with the beta version of the software.
Initial benchmarks for beta GATK4
The Whole Genome Pipeline enables identification of variants from a human whole-genome resequencing experiment using the best practices workflow for alignment and variant calling (Figure 1). The pipeline is designed for optimum performance on data from experiments that use a PCR-free library preparation protocol and targets 30x mean coverage across the genome, but is suitable a range of coverages (verified up to 150x).
As with all bioinformatics workflows on our Platform, we extensively benchmarked the GATK4 whole genome workflow to ensure suitability and performance across multiple analysis scenarios.
1 Precision and recall
We use the Genome In A Bottle truth dataset and the hap.py variant evaluation tool to measure the workflow sensitivity and precision for both SNPs and indels following the GA4GH recommended practices (Table 1). We note that the observed precision and sensitivity measures are in line with expected performance for this sample. Further, we observe that both the transition to transversion ratio for SNPs as well as the ratio of heterozygous to homozygous indels calls are in the expected ranges for a whole-genome context. Only the calls that have PASS flag in the VCF filter column were used for calculating the reported performance metrics, reflecting the performance that can be expected when the pipeline is used in a production scenario.
| Sample | SNP precision | SNP sensitivity | Indel precision | Indel sensitivity | SNP ti/tv-ratio | Indel het/hom-ratio |
| HG001 | 99.8 % | 99.2% | 87.3% | 91.5% | 2.11 | 1.8 |
Table 1 | Precision and recall for the beta GATK4 workflow using the NA12878 (HG001) sample. In addition we report the observed ratio of transitions to transversions for SNPs and homozygous to heterozygous indels. All metrics are in line with expected performance for this sample.
2 Mendelian consistency
Second, we use the CEPH and Ashkenazi trios from the Genome In A Bottle project to measure pipeline genotyping consistency. This is done by counting the total proportion of fully determined variant loci where the detected trio genotypes for father, mother, and child agree with the expected mendelian inheritance pattern (Table 2). For the two trios we observe SNP error rates of <1% and indel error rates of 6.38% and 3.85%, respectively.
| Pedigree | Total SNP calls | Total indel calls | SNP error rate | Indel error rate |
| CEPH | 4836659 | 933789 | 0.82% | 6.38% |
| Ashkenazi | 4908585 | 1056532 | 0.62% | 3.85% |
Table 2 | Mendelian consistency. The consistency of the of detected pipeline genotypes as measured by compliance with Mendelian inheritance. The higher error rates for the CEPH trio reflect the slightly higher quality of the Ashkenazi dataset.
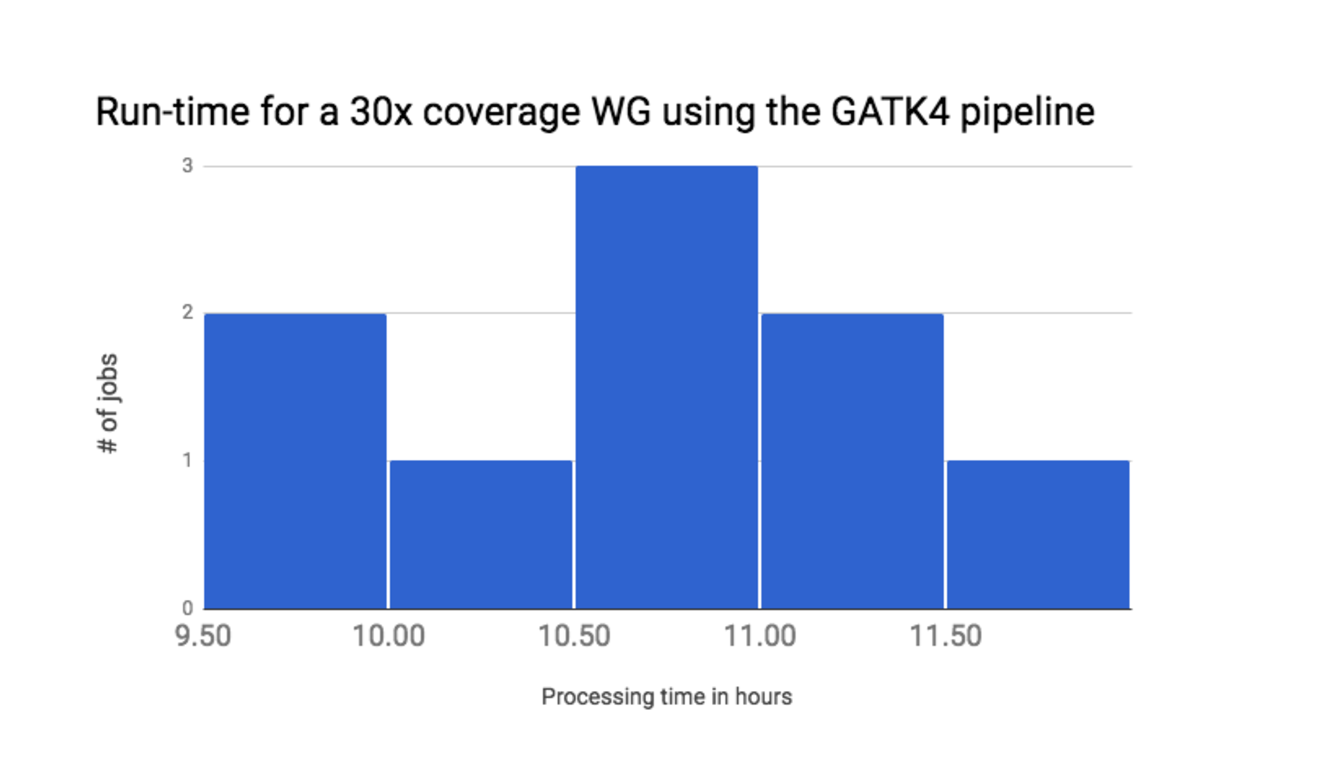
3 Run time
Finally, we measured the end-to-end run-time of the workflow. The average run-time for analyzing 10 whole-genome sequencing experiments on default AWS hardware is around 10.5 hours for a 30x PCR-free sample (Figure 2). These run-time results already constitute a 30% improvement relative to our GATK3.7 pipeline. We expect to see further run-time improvements as we work towards a pipeline for the official GATK4.0 release.
Running GATK4 on Seven Bridges
Researchers can use the Seven Bridges Platform to quickly deploy the latest beta version of the GATK4 workflow for variant calling. With the transition to the open source licence, there is no need for researchers in start-ups, biotech or pharma to obtain a separate licence to use to software. As this whole-genome workflow uses the beta-2 version of the GATK v4.0 suite, we recommend careful review of the git repository notes before using this workflow for production work.
We will offer access to the full GATK4 release when it becomes available later this year. Stay tuned for more posts on our work benchmarking, optimizing and enhancing open source GATK4 to help researchers extract maximum value from their research data.
Seven Bridges bioinformaticians constantly work to curate and optimize community standard bioinformatics workflows, such as open source GATK4. Speak to our scientists to see how our team can help you increase speed, reduce costs and enhance functionality of your investigational and production workflows.