The representation of the human reference genome as a linear haploid DNA sequence poses limitations when trying to incorporate the known genetic diversity of human populations. This has led to the development of graph-based references, able to naturally represent all polymorphisms, including insertions, deletions, and structural variation.
Seven Bridges GRAF™ Suite comprises bioinformatics workflows and tools for secondary analysis of next generation sequencing (NGS) data, based on a pan-genome graph reference. These tools are able to call variants with superior accuracy, without compromising on speed or cost. GRAF™ uses standard genomic data formats (FASTA, VCF, FASTQ, BAM, CRAM, BED), ensuring compatibility with existing workflows. All Seven Bridges GRAF™ workflows are specified in CWL 1.0, ensuring portability across various compute environments.
Seven Bridges GRAF™ enables:
- Large human population studies by using a population-specific genome graph comprising millions of individuals
- Precise analysis of individual genomes by making use of personalized genome graphs and family genome graphs leading to accurate de novo mutation detection
- Rare disease studies by using a curated graph containing mutations associated with the disease
Seven Bridges GRAF™ Suite consists of:
Workflows
- GRAF™ Germline Variant Detection Workflow – uses a Pan-Genome graph containing genetic information from many populations around the world. The workflow accepts input reads in FASTQ, CRAM or BAM format, produces graph-based alignments in BAM or CRAM format, and outputs variant calls in VCF format. In this blog post, we discuss some of the fundamental advantages of our Pan-Genome approach compared to other linear or graph-based methods.
- GRAF™ Extended Germline Variant Detection Workflow – makes use of an ancestry-aware genome graph (i.e. a population-specific reference) in order to increase sensitivity towards the population represented by the graph reference. The workflow also contains a downstream supplementary part that merges and annotates all variants and outputs a single multi-sample VCF file. The ancestry-aware approach constitutes a tailored approach for targeted population studies. The results for the African population showcasing the benefits of this approach can be found in our most recent blog post and pre-print.
Graph References
- GRAF™ Pan Genome Reference – contains both small variants (SNPs and INDELs up to several dozens of base-pairs in length) as well as larger structural variants, which are typically difficult to identify from short-read sequencing data. Consequently, graph references provide the means to both accurately detect structural variation and thus reduce spurious variant calls.
- GRAF™ Admixed American Reference – curated from a set of 8,000 samples from various public databases covering the whole genome. This graph is augmented with population-specific structural variants obtained from PacBio HiFi reads.
- GRAF™ African Reference – curated from a set of more than 20,000 samples collected from various public databases covering the whole genome. This graph is augmented with population-specific structural variants obtained from PacBio HiFi reads.
- GRAF™ East Asian Reference – curated from a set of more than 3,000 samples collected from various public databases covering the whole genome. This graph is augmented with population-specific structural variants obtained from PacBio HiFi reads.
- GRAF™ European Reference – curated from a set of more than 34,000 samples collected from various public databases covering the whole genome. This graph is augmented with population-specific structural variants obtained from PacBio HiFi reads.
- GRAF™ South Asian Reference – curated from a set of approximately 3,000 samples collected from various public databases covering the whole genome. This graph is augmented with population-specific structural variants obtained from PacBio HiFi reads.
Services
- GRAF™ Construction Service – consists of bioinformatics methods and services that enable the construction of representative and accurate genome graph references. The intended use case for such tailored graphs can range from pan-genome and population studies to personalized or disease related applications.
Tools
- GRAF™ Aligner – maps sample reads against a Genome Graph Reference, implicitly considering many alternate haplotypes at each locus, thereby minimizing reference bias.
- GRAF™ Variant Caller – enables integrated calling of SNPs and INDELs, as well as structural variants present in the Genome Graph.
- GRAF™ Stats – calculates graph-specific alignment metrics and measures the utilization of the graph reference in read alignment, in addition to standard metrics such as coverage, unmapped, multi-mapped read counts and improper alignments.
- GRAF™ Genome Viewer – a genome viewer that can visualize graph references and alignments made against the edges of a graph reference.
Genome Graph is a powerful genomic data structure
Seven Bridges GRAF™ Suite utilizes advanced data structures to generate directed acyclic graphs and is a fundamental rethinking of how genomic variation data is represented. Unlike standard linear references, this structure makes use of information from an entire population to characterize genetic variants with unprecedented accuracy. Graph structures have the potential to learn from every new individual sequenced, meaning that the graph-based reference can improve with each additional genome. Better still, this improvement happens with only minimal increases in file size, allowing analysis of genomes at population scale.
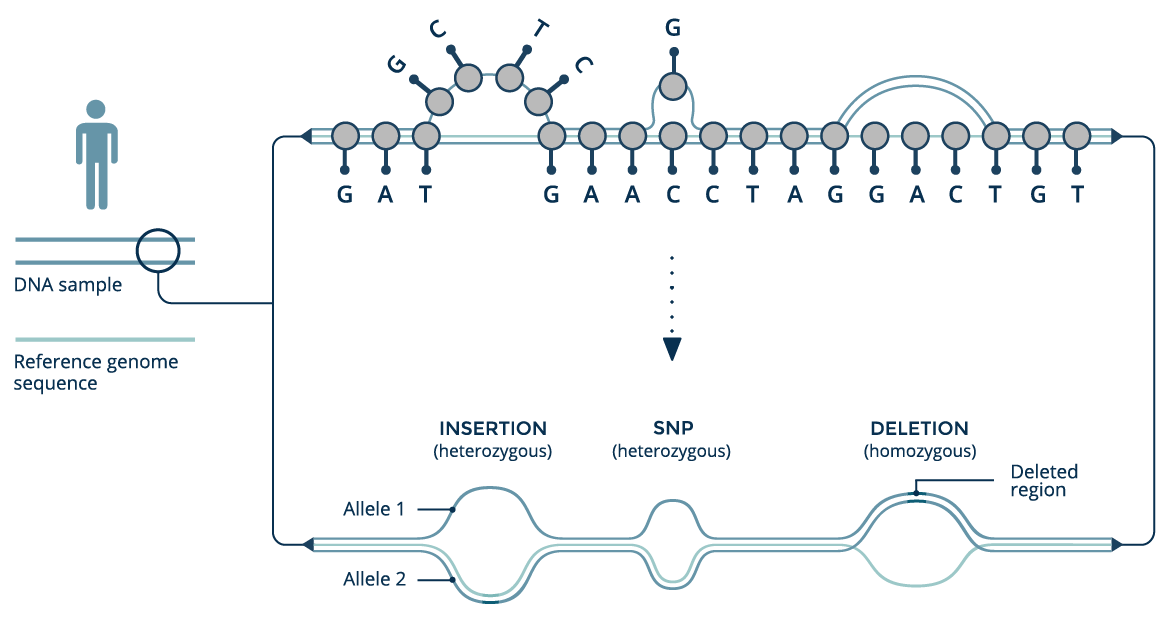
The GRAF™ Pan Genome Reference organizes genomic data from a population into an edge-based sequence variation graph. In such graphs, edges are the primary data carrier elements and alternative haplotypes are represented as different paths through the graph. The linear reference assembly forms the graph backbone, and additional variants are added as new edges in the graph. A longer genomic haplotype can be obtained by following a path through the graph and concatenating the (sub)sequences contained by the visited edges. We call this structure a genome graph.
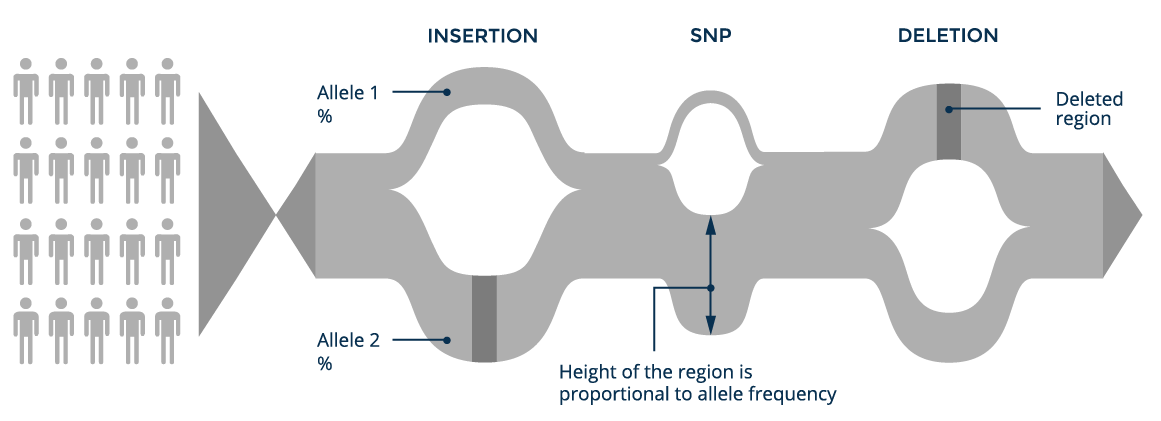
A graph genome reference can contain both small variants (SNPs and indels up to several dozen base-pairs in length) and the larger structural variants, which are typically difficult to identify using short-read sequencing technologies. Consequently, graph references provide a framework to both accurately detect structural variation and reduce the error rate in calling small variants.
Find out how to build a better reference genome
Graph structures enable accurate variant calling
Seven Bridges GRAF™ Suite enables accurate variant calling — the process of determining the set of genomic variants present in a sample. Current state-of-the-art variant callers are based on local reassembly of reads mapped in the region surrounding a putative variant. This approach significantly improves the accuracy of variant calls but is limited by the choice of size of the reassembly region, and the size of the expected assembled contig. If these values are set too small, variants can be missed. If these values are set too big, it can be computationally prohibitive.
Using GRAF™ can alleviate these issues, as reads which are partially aligned to a known variant edge allow for better adaptive selection of the necessary reassembly window as the edge from a graph reference contains information regarding its size. Furthermore, GRAF™ enables a completely different paradigm in variant calling — for variants present in the graph, sequence and the location information is known, thus it is possible to genotype directly against the graph.
The GRAF™ Aligner utilizes a pan-genome graph structure, created by augmenting the reference genome with a set of known variations, significantly reducing the effects of reference bias. The graph genome incorporates variants from a variety of sources, including the 1,000 Genomes Project and the Simons Genome Diversity Project. By preserving all of the variant information, GRAF™ facilitates better alignment than linear alternatives, resulting in more accurate variant calls.
Learn more about aligning reads to a reference graph
Resources