Seven Bridges is at the BioIT World Conference & Expo showcasing how we’re helping researchers do more with their biomedical data. Our Platform, Cavatica, Sonar and the Graph Genome Suite support secure, scalable and collaborative research. Set up a meeting with us and join us for the following presentation:
Graph Genome Tools for Precision Medicine
 May 24 | 12pm
May 24 | 12pm
Next-Gen Sequencing Informatics & Cancer Informatics Tracks
Jack DiGiovanna, Director of Program Management
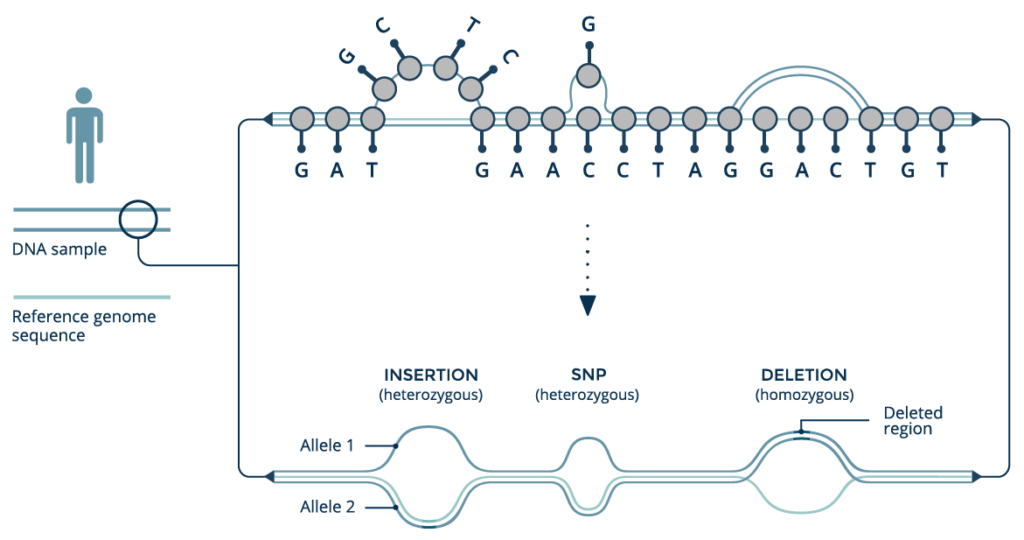
An emerging paradigm of graph-based reference genome structures represents a fundamental rethinking of how genomic variation impacts health. Unlike standard linear references, graph genomes can use information from an entire population to characterize genetic variants with unprecedented accuracy, improving as new genomes are added to the graph structure. Graph genomes improve accuracy across myriad classes of genetic variation, including structural variation, and are ideal for analyzing highly variable loci and sequences from underrepresented populations. We will discuss graph genomes in general and describe their implications for the future of precision medicine.
Rabix: An Open-Source Toolkit for the Composition and Execution of Reproducible, Portable and Scalable Bioinformatics Workflows
 May 23 – 25 | Poster Session
May 23 – 25 | Poster Session
Adrian Sharma, Program Manager
Biomedical data analysis workflows created using ad hoc methods are not easily sharable or reproducible, which can result in inconsistencies when attempting to replicate scientific results and publications or perform collaborative analyses. To overcome these challenges, the bioinformatics community is embracing the use of software containers such as Docker and workflow description languages for easier documentation and replication of analysis pipelines. Software containers enable easy packaging and sharing of command line tools and bioinformatics applications. However, containers do not come with instructions on how to execute these applications in a fashion that is human and machine-readable, enabling automation, scaling and reproducibility. The Common Workflow Language (CWL), established in 2014, is a community-developed specification for describing containerized tools and workflows for such a purpose. CWL is written in easily readable and parsable YAML, and also allows for extensions and tooling that facilitate code development, testing, and execution.
One such set of tools our Rabix project, an open-source project that enables rapid composition and execution of CWL workflows in a manner that is scalable and reproducible. CWL is intended to be a comprehensive, deep-level description of analyses; therefore, it can be very verbose and time-consuming to compose by hand, particularly for common bioinformatics applications such as whole genome or exome sequencing. Internal and community feedback demonstrated that CWL workflows can be both time-consuming and challenging to compose due to the sheer number of tool inputs, outputs, parameters, and inter-tool connections. To improve developer productivity, we developed the Rabix Composer, a standalone integrated development environment that provides rich visual and text-based editors for the creation of tools and workflows in CWL. The Rabix Composer allows developers to easily develop tools in teams, integrate with services like GitHub, synchronize tools with cloud platforms like Seven Bridges, view documentation, request support from the developer team, annotate their CWL workflows, and track version history, from a single interface. Parsing and executing CWL descriptions across various compute environments can also be challenging.
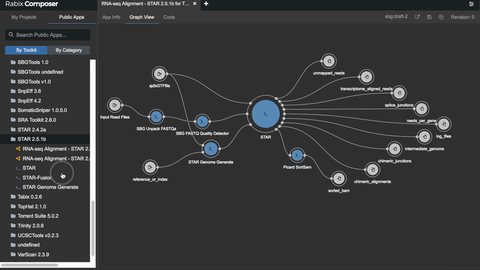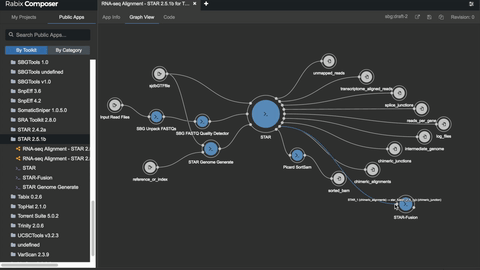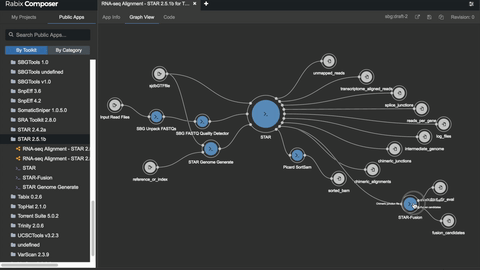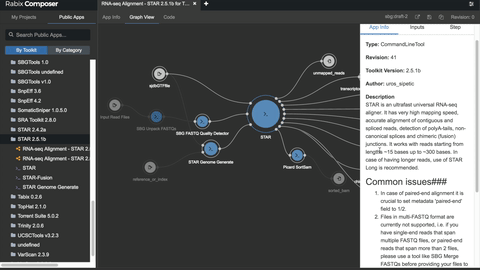
We developed an implementation of CWL, the Rabix Executor, which allows for the execution of CWL on single machines (e.g. OS X, Linux), HPC, or multiple cloud environments. The Rabix Composer and Executor enhance the CWL user experience by providing task and error logs, the ability to restart workflows upon failure, the ability to implement custom interfaces (e.g. command line interface, graphical user interface), support for multiple versions of CWL within a single workflow, backwards compatibility for previous tools and workflows, and optimizations for computation such as batch processing, parallelization and improved job scheduling efficiency. In sum, the Rabix project enhances data analysis by improving reproducibility, simplifying software sharing/publication, and reducing the friction when attempting to use a new algorithm or replicate a scientific finding.
Enabling Scalable and Rapid Metagenomic Profiling of the Transcriptome with the Seven Bridges Cancer Genomics Cloud
 May 23 – 25 | Poster Session
May 23 – 25 | Poster Session
Raunaq Malhotra, Bioinformatics Scientist
The advent of next-generation sequencing has accelerated the generation of genomic data and created a need for methods to organize, share, and analyze large volumes of data. To date, petabytes of multidimensional information from thousands of patients have been collected. Access and analysis of this information becomes increasingly challenging as the volume and heterogeneity of the data increases. This difficulty is exemplified when we consider data generated by the efforts of The Cancer Genome Atlas (TCGA) network, which encompasses more than 2.5 petabytes. Historically, downloading the complete TCGA repository can take weeks to months. Further large-scale reanalysis is limited to researchers with access to large institutional compute clusters. In addition, maintaining and synchronizing multiple copies of the TCGA dataset incurs additional costs. The goal of the National Cancer Institute’s (NCI) Cancer Cloud Pilot project is to democratize access to TCGA by co-localizing data with the computational resources to analyze it. The project was born out of the recognition that biological research is becoming increasingly computationally-intensive and new approaches are required to support effective data discovery, storage, computation, and collaboration.
Funded by the NCI, the Seven Bridges Cancer Genomics Cloud (CGC) enables researchers to leverage the power of scalable cloud computing to gain actionable insights about cancer biology and human genetics from large public datasets including TCGA, the Simons Genome Diversity Project dataset, and the Cancer Cell Line Encyclopedia. Our approach is to create a secure, cloud-based cancer research platform that includes collaborative tools for data upload, analysis, and visualization. Our solution uses resource description frameworks, data harmonization, and metadata curation to facilitate effective querying. Additionally, we implemented the Common Workflow Language, an emerging standard for describing computational workflows, to support computational reproducibility.
Since its launch in February 2016, more than 1500 researchers have used CGC to access and analyze more than 50,000 TCGA samples. Our presentation will describe the motivation, inception and development of the CGC, and include a case study on the application of metagenomic profiling tools to identify viral transcripts using RNA sequencing data from TCGA. Viral transcripts can act as markers of cancer incidence, e.g.HPV and cervical cancer, and the CGC allows us to profile a cohort of 300 samples for less than $20 and in 8-10 hours; this time and cost are much less than it would have taken without the CGC. We will demonstrate how cloud computing resources can facilitate the interrogation of viral transcripts in TCGA and other data using publicly-available or custom pipelines.
Visit Us in the Exhibit Hall
We’re at booth 432 discussing how we help researchers do more with their biomedical data. Come by to see how our tools open up massive biomedical datasets for exploration and put fully reproducible, scalable genomic analysis into the hands of anyone with an internet connection.
- Tuesday, May 23 | Exhibits Open 5pm – 7pm
- Wednesday, May 24 | Exhibits Open 9:50am – 6:30pm
- Thursday, May 25 | Exhibits Open 9:45am – 1:55pm