Neoantigen Discovery Using Next-Generation Sequencing Data
Update (5/26/20) – Since originally publishing this post we’ve hosted a webinar on our Neoantigen Workflow. If you’d like an even deeper look at how to harness your NGS data, just click here!
Leveraging neoantigens for guided treatment has become the definition of personalized cancer immunotherapy. Given their potential to eradicate nonspecific treatments such as chemotherapy, a host of therapeutic studies are underway to further understand how neoantigens can be used to trigger immunity-induced tumor regression. Seven Bridges has developed a workflow for neoantigen discovery using next-generation sequencing (NGS) data, which analyzes tumor-normal pairs of whole exome sequencing (WES) samples and tumor RNA-seq gene expression data in order to output candidate epitopes for neoantigens.
This neoantigen discovery workflow has been tested throughout the last three rounds (Round 2 in 2017, Round X and Round 3 in 2019) of the Tumor Neoantigen Selection Alliance (TESLA) Challenge organized by the Parker Institute for Cancer Immunotherapy. The TESLA Challenge has gathered more than 35 leading neoantigen research groups in academia and industry with the goal of validating neoantigen prediction workflows. For Round X, TESLA provided flow-cytometry-validated peptide-HLA sets for approximately a dozen patients. The neoantigen discovery workflow developed by Seven Bridges managed to detect the majority of confirmed candidates. Detailed results of the challenge will soon be published by the TESLA organizers.
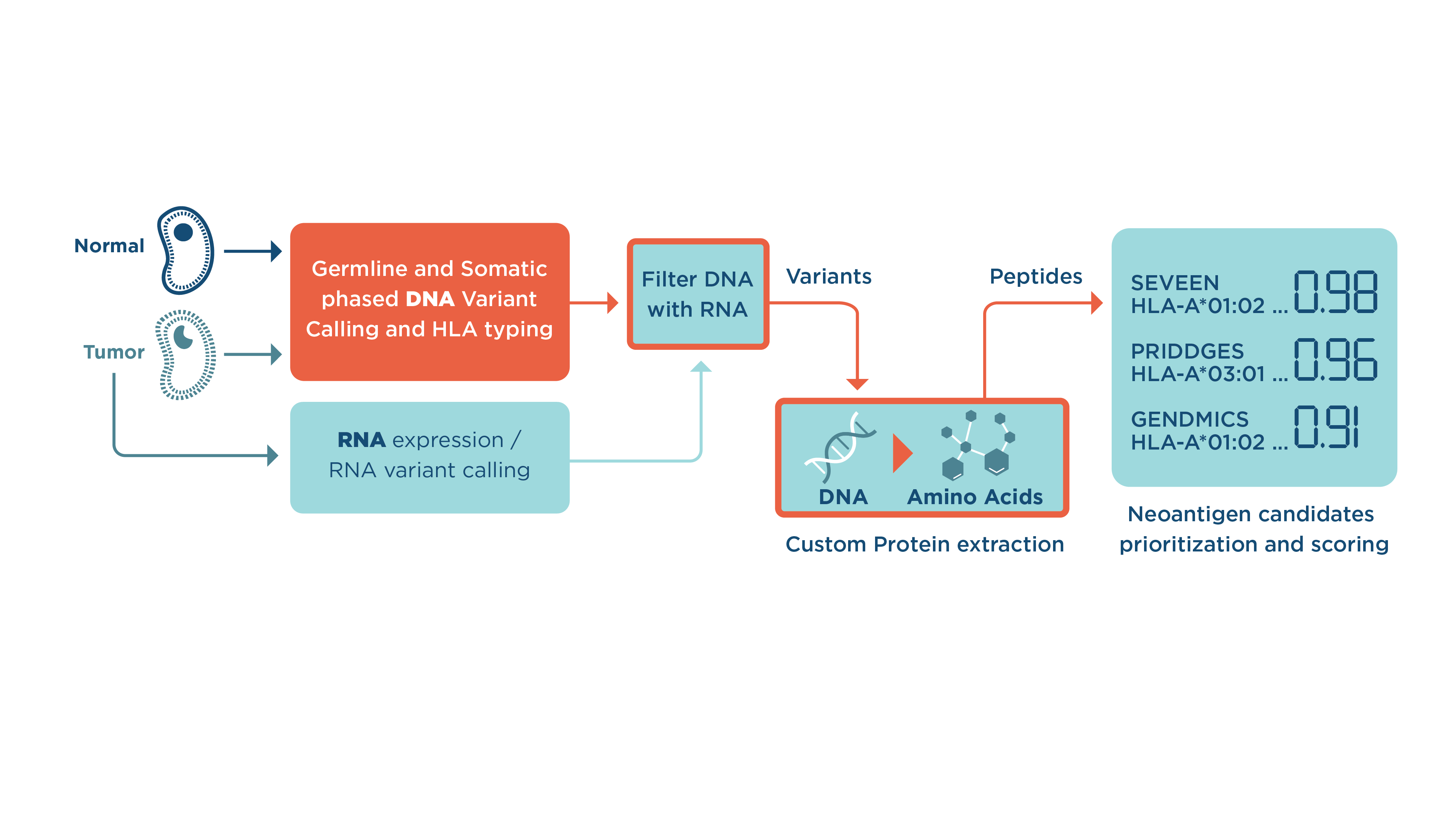
- First, the workflow will perform read alignment, with the preparation of aligned files, leveraging GATK best practices.
- Next, somatic variant calling (using Strelka2 and Mutect2) is performed and variants are merged into one file (by selecting intersect or union).
- Once the variants are merged into one file, variant phasing occurs. Variant phasing is the separation of variants belonging to the same chromosome (Figure 2). The purpose of this step is to accurately reconstruct the nucleotide sequence which is translated into the neoantigen candidate.
- Now you are ready to move onto the following three steps that occur in parallel with the alignment, somatic variant calling, and variant phasing:
- RNA reads are processed with Salmon and RSEM tools, which enable high precision quantification of transcripts. The output from this part is a list of gene isoforms with corresponding RNA expression scores.
- GATK4 RNA Variant Calling which outputs tumor RNA variants.
- Optitype calculates Human Leukocyte Antigen (HLA) types from tumor FASTQs.
Two close variants: Chr5 20000 C T 0/1 Chr5 20011 A G 0/1 Which two alleles will exist? ...C...A… ...C...G… ...T...A… ...T...G… Phasing will tell us! After phasing: Chr5 20000 C T 0|1 Chr5 20011 A G 1|0 Two alleles will be called: ...C...G… and ...T...A… (...C...A… and ...T...G… will not be considered)
- Next, phased somatic DNA variants, optionally filtered with RNA expressed regions (to boost-up precision of neoantigen candidates), are passed to protein extraction tools to extract the nucleotide sequences around the positions of the somatic variants and translate them to proteins. During this process, the protein extraction tools take into account the full range of complex changes which are currently not existent within the published workflows: changes from both germline and somatic variants (SNPs and indels), mutations (SNPs or indels) within the STOP codon (nonstop), and mutations that create STOP codons (nonsense). The extracted protein sequence and identified HLA type are used by the epitope prediction tools (NetCTLpan, IEDB Processing: NetMHCPan, Pickpocket, etc) to compute confidence-ranked epitope candidates.
- Finally, a list of the confidence-ranked neoantigen candidates with HLA types, variant information, RNA expression, and variant calling scores are merged and prioritized in the analysis epitopes tool. Its output is shown in Table 1.
| Sequence name | HLA Type | Peptide | NetMHC Score | Pickpocket Score | NetCTLPan Combined Score | Transcripts per Million (RNA) | Variant Information [GT:AD:AF:…]|transcript_ids] |
|---|---|---|---|---|---|---|---|
| 1_111957245_C_A | HLA-A*02:01 | MMLSSSPV | 0.881 | 0.633 | 1.09815 | 11.5 | 0/1:261 4:0.018:2|ENST00000341809|ENST00000553168 |
| 8_144392368_T_C | HLA-A*02:01 | WLLEKLEQL | 0.828 | 1.097 | 1.06133 | 12.5 | 0/1:103 12:0.091:6:6:|ENST00000533313|ENST00000332271|ENST00000527701 |
| 17_28537638_C_T | HLA-A*02:01 | VLDEFPHV | 0.836 | 0.374 | 1.06015 | 23 | 0/1:94 4:0.048:|ENST00000579221|ENST00000394821 |
| 1_111957245_C_A | HLA-A*02:01 | MMLSSSPVIQL | 0.816 | 1.273 | 1.05881 | 6.7 | 0/1:261 4:0.018:|ENST00000553168 |
| 9_32630149_G_T | HLA-B*07:02 | RPKQHFML | 0.776 | 0.901 | 1.01862 | 9.6 | 0/1:312 27:0.083:13|ENST00000242310 |
| 11_58207203_C_T | HLA-A*02:01 | YLAIGSYI | 0.793 | 0.705 | 1.01312 | 6 | 0/1:67 6:0.076:|ENST00000302572 |
Table 1: Example output of scored sorted neoantigen candidates with all necessary information
Non-proprietary components of this portable and reproducible workflow will be publicly available on the Seven Bridges Platform. They are described in Common Workflow Language (CWL) to ensure portability and reproducibility between computation environments. Execution times of the workflow per patient varies between 3 and 6 hours ($2.3 to $4.5), depending on the type of tumor and mutation burden.
Combining this workflow with the automation, customization and scalability of the Seven Bridges Platform enables rapid prioritization of a patient’s neoantigen candidates